
N-methyl-D-aspartate receptor functions altered by neuronal PTP1B activation in Alzheimer’s disease and schizophrenia models
2022-03-09 07:22AlexandreStewartHsiaoHueiChen
中国神经再生研究(英文版) 2022年10期
Alexandre F. R. Stewart, Hsiao-Huei Chen
Glutamate is the main excitatory neurotransmitter in the brain and binds to two major classes of receptors, the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and the N-methyl-D-aspartate (NMDA) receptors. Unlike AMPA receptors that are immediately activated by glutamate release, NMDA receptors are blocked by magnesium and can only be activated by glutamate after membrane depolarization. Thus, NMDA receptors are only activated after repeated AMPA receptor activation by glutamate.NMDA receptors are, for the most part,calcium-permeable channels. Calcium influx through NMDA receptors modulates synaptic transmission in neurons based on prior history of excitation, and provides a means of scaling the strength of synapses required for Hebbian plasticity. NMDA receptors were first characterized in the post-synaptic membrane, where calcium influx controls AMPA receptor levels and activity-dependent gene expression.Tyrosine phosphorylation of postsynaptic NMDA receptors promotes calcium influx,whereas dephosphorylation of NMDA receptors causes their internalization and reduces calcium influx through NMDA receptors (Wang and Salter, 1994). More than 10 years ago, the striatal-enriched tyrosine phosphatase Step61 was tied to NMDA receptor dephosphorylation in the context of Alzheimer’s disease and amyloid-beta (Kurup et al., 2010).
In recent years, NMDA receptors have also been found in the presynaptic membrane, where calcium influx regulates neurotransmitter release,including glutamate. Moreover, NMDA receptors signal not only as calcium channels (an ionotropic function), but other cell signaling mechanisms involving protein kinases and phosphatases(a metabotropic function) has also emerged. Recent reports have highlighted the functional consequences of the activation of the tyrosine phosphatase PTP1B in glutamatergic neurons on two NMDA receptor-dependent functions.In one study, the cellular mechanism of defective plasticity at the CA3:CA1 synapse underlying cognitive deficits in a mouse model of Alzheimer’s disease were revealed as a deficit in presynaptic NMDA receptors that could be fully restored by neuronal ablation of PTP1B or by the selective PTP1B inhibitor Trodusquemine(Zhang et al., 2021). In a second study,the schizophrenia-like effects of the NMDA receptor antagonist ketamine were shown to depend on PTP1B. Neuronal ablation of PTP1B or PTP1B inhibition with Trodusquemine prevented these schizophrenia-like functional deficits (Qin et al., 2021).
The tyrosine phosphatase PTP1B is expressed in neurons of the hypothalamus,the hippocampus, and cerebral cortex.Normally, PTP1B is maintained in a quiescent state by a selective interaction with small protein inhibitors, including the LIM-only domain protein LMO4 (Pandey et al., 2013). Under physiological conditions,PTP1B is only transiently activated to limit signaling by tyrosine kinase receptors.However, emerging evidence indicates that several cellular stresses including dietary stress (Pandey et al., 2013), hormonal stress (Qin et al., 2015), tissue injury, and misfolded protein aggregates (Ricke et al.,2020) constitutively activate this tyrosine phosphatase causing deficits in cellular signaling by the insulin receptor (Pandey et al., 2014), the leptin receptor (Zhou et al., 2012), and the BNDF receptor trkB (Qin et al., 2020), to name a few.
In a mouse model of congenital Alzheimer’s disease, the hAPP-J20 mouse that expresses a mutant form of human β-amyloid precursor protein, neuronal PTP1B is activated and hastens the onset of cognitive and spatial memory deficits(Ricke et al., 2020). Systemic inhibition with the selective PTP1B non-competitive allosteric inhibitor trodusquemine, a compound that crosses the blood-brain barrier, ameliorated cognition and spatial memory in the Morris water maze test,preventing inflammation and neuron loss at the CA3 region of the hippocampus of hAPP-J20 mice. Similarly, hAPP-J20 mice in which PTP1B was selectively ablated in glutamatergic neurons were spared cognitive and spatial memory deficits and neuron loss at the CA3 region (Ricke et al., 2020) despite no marked reduction in inflammation compared to hAPP-J20 littermates. While this study pointed to a critical involvement of neuronal tyrosine phosphatase PTP1B in the progression of Alzheimer’s-like deficits in these mice,the functional deficits at the cellular level remained unknown.
The hippocampus is a brain region important for learning and remembering spatial cues and long-term changes in the strength of synapses between neurons of the hippocampus are thought to underlie the formation of spatial memories. A follow-up study by Zhang et al. (2021)examined synaptic strength and plasticity from CA3 neurons onto CA1 neurons of the hippocampus to identify cellular mechanisms disrupted by neuronal PTP1B activation in the hAPP-J20 mouse.Given that hAPP-J20 mice fail to acquire spatial memories, hippocampal synaptic plasticity was likely compromised in these mice. Zhang et al. (2021) found a marked deficit in long-term potentiation(LTP) of the CA3:CA1 synaptic response that was rescued by systemic PTP1B inhibition or genetic ablation of PTP1B specifically in glutamatergic neurons(Figure 1A). Bath application of the NMDA receptor blockers MK801 or D-2-amino-5-phosphonopentanoate to wild-type hippocampal brain sections phenocopied the deficit in LTP seen in hAPP-J20 mice and reversed the benefit of glutamatergic neuron-specific ablation of PTP1B in hAPP-J20 mice. Thus, NMDA receptors were clearly implicated in the process.However, given that both CA3 and CA1 neurons are glutamatergic, it was not clear whether PTP1B ablation rescued a pre- or post-synaptic deficit in hAPP-J20 mice.
Surprisingly, the inclusion of the membrane-impermeable NMDA receptor blocker norketamine in the postsynaptic recording electrode silenced post-synaptic NMDA receptors, but had little effect on LTP in either wild type mice or in hAPP-J20 mice rescued with glutamatergic neuron-specific ablation of PTP1B, strongly supporting a pre-synaptic deficit in NMDA receptor-dependent LTP.Using the coefficient of variation method,Zhang et al. (2021) further confirmed that the deficit in NMDA receptordependent LTP at CA3:CA1 synapses is largely presynaptic in hAPP-J20 mice. This result is important, because it suggests the expression of mutant human amyloid-beta protein constitutively activates PTP1B in presynaptic terminals, leading to a deficit in NMDA receptor-mediated synaptic plasticity and learning deficits in these mice. It should be pointed out, however,that the study by Zhang et al. (2021) used a paired stimulation protocol that favors presynaptic LTP and did not address a postsynaptic effect of PTP1B in hAPP-J20 mice.Future studies using different stimulation protocols that induce LTP mainly through postsynaptic NMDA receptors will be needed to adequately address this question. Especially, whether PTP1B works through Step61 to affect postsynaptic NMDA receptors in the context of amyloidopathy (Kurup et al., 2010). It is also worth noting that Thompson’s group recently reported another mechanism whereby post-synaptic NMDA receptors modulate presynaptic glutamate release via a retrograde signaling mechanism.Unlike Zhang et al. (2021) who reported a direct effect on pre-synaptic NMDA receptors to regulate glutamate release,Bialecki et al. (2020) showed that postsynaptic metabotropic NMDA receptors activate Src kinase to modulate postsynaptic pannexin-1 (Panx1) channeldependent production of anandamide (an endovanilloid). Anandamide retrogradely activates pre-synaptic TRPV1 (transient receptor potential vanilloid 1) receptors that suppress glutamate release (Bialecki et al., 2020).
Zhang et al. (2021) also reported reduced phosphorylation at tyrosine 1472 of the GluN2B subunit of the NMDA receptor that was restored by Trodusquemine or glutamatergic neuron-specific ablation of PTP1B, while phosphorylation of tyrosine 1325 or the GluN2A subunit was unaffected. At first glance, this result suggests that dephosphorylation of presynaptic GluN2B rather than GluN2A subunits are chiefly responsible for the LTP deficit. However, GluN2A is phosphorylated at tyrosine 934 and Zhang et al. (2021) did not interrogate this site using a specific antibody(ThermoFisher PA5-105627) that recently became available. What is clear is that either pharmacological inhibition of PTP1B or genetic ablation of PTP1B in glutamatergic neurons restores NMDA receptor-dependent LTP at the CA3:CA1 synapse in this mouse model of hereditary Alzheimer’s disease with amyloidopathy.Pressing questions are whether other models of Alzheimer’s disease (for example, models with tauopathy like the PS19 mice expressing human mutant Tau P301S protein) will show a similar response to PTP1B inhibition or ablation and whether sporadic Alzheimer’s disease will respond to therapeutic interventions targeting PTP1B.
PTP1B hyperactivity is clearly deleterious to NMDA receptor signaling and synaptic plasticity important for spatial memory and learning in Alzheimer’s disease.Another recent study reported that PTP1B hyperactivity is implicated in schizophrenia-related deficits that are also tied to impaired NMDA receptor function. Subanesthetic doses of the NMDA receptor antagonist ketamine produce several schizophrenia-like behaviors in mice, including deficient sensorimotor gating of the acoustic startle response, spatial learning deficits, and hypermobility. These behavioral effects of ketamine can all be reversed by the PTP1B inhibitor Trodusquemine, while glutamatergic neuron-specific ablation of PTP1B prevents the sensorimotor gating and spatial learning deficits, but not the hyperlocomotion (Qin et al., 2021).Thus, PTP1B activation in glutamatergic neurons contributes to sensorimotor gating and spatial learning deficits caused by subanesthetic doses of ketamine,whereas PTP1B activation in nonglutamatergic neurons is responsible for the hyperlocomotion effect of ketamine.Moreover, this study suggests that PTP1B activation is a consequence of silencing NMDA receptors with ketamine (Figure 1B).
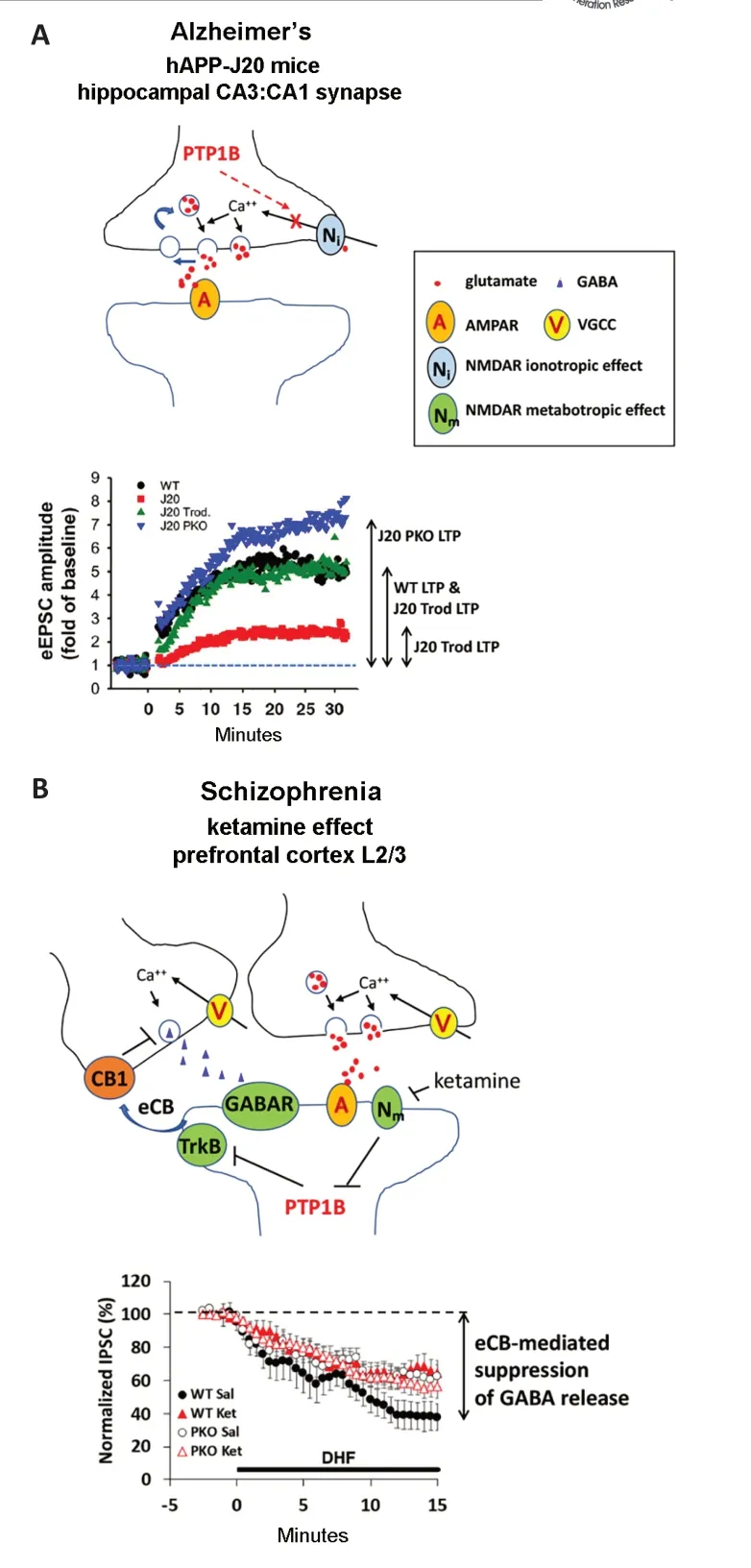
Figure 1 |PTP1B activity impedes NMDA receptordependent pre- and post-synaptic functions.(A) The diagram on top illustrates the effect of PTP1B activation to inhibit presynaptic NMDA receptor ionotropic function required for LTP. Long-term potentiation (LTP) of the amplitude of evoked excitatory postsynaptic currents(eEPSC) at the CA3:CA1 synapse of the hippocampus is impaired in hAPP-J20 Alzheimer’s disease mice due to hyperactivation of PTP1B that prevents NMDA receptordependent augmentation of excitatory neurotransmitter release (Zhang et al., 2021). LTP in hAPP-J20 mice is restored by chronic Trodusquemine administration.Genetic ablation of PTP1B in glutamatergic neurons(PKO) increases LTP in hAPP-J20 mice relative to WT.Both pharmacological inhibition and genetic ablation of PTP1B prevent cognitive decline and neuron loss in hAPP-j20 mice. (B) The diagram on top suggests a metabotropic effect of NMDA receptors to suppress PTP1B is blocked by ketamine. Ketamine, an NMDA receptor antagonist, impairs endocannabinoid (eCB)mobilization in response to the trkB receptor agonist 7,8-dihydroxyflavone (DHF) through disinhibition of PTP1B in layer 2/3 pyramidal neurons of the prefrontal cortex(Qin et al., 2021). trkB activation mobilizes eCB that acts on presynaptic CB1 receptors on inhibitory neurons and reduces the amplitude of inhibitory (GABA) post-synaptic currents (IPSC) recorded at layer 2/3 pyramidal neurons Schizophrenia-like effects of ketamine are blocked by pharmacological inhibition or genetic ablation of PTP1B.Data shown in A and B are modified from studies by Zhang et al. (2021) and Qin et al. (2021), respectively. AMPAR:α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor; GABA: γ-aminobutyric acid; IPSC: inhibitory postsynaptic currents; J20: hAPP-J20; NMDAR: N-methyl-Daspartate receptor; VGCC: voltage-gating calcium channel;WT: wild type.
Intriguingly, a genetic manipulation that removed the endogenous inhibitor of PTP1B (i.e., LMO4) in glutamatergic neurons also produced a mouse model with constitutively active PTP1B that resulted in schizophrenia-like deficits in sensorimotor gating and spatial memory, but not hyperlocomotion. A consequence of PTP1B activation in post-synaptic glutamatergic neurons is dephosphorylation and inactivation of the trkB receptor for brain-derived neurotrophic factor, a growth factor released from neurons in an activitydependent manner. In layer 2/3 pyramidal neurons of the medial prefrontal cortex, trkB receptors activate phospholipase C and contribute to the production of the endogenous cannabinoid 2-arachidonoylglcerol that suppresses γ-aminobutyric acid release from presynaptic inhibitory neurons.Functionally, sensorimotor gating and spatial memory deficits were correlated with loss of trkB receptor-dependent endocannabinoid mobilization to modulate inhibitory inputs to layer 2/3 pyramidal neurons (Qin et al., 2020). Importantly,these cellular signaling deficits and schizophrenia-like behaviors could be fully rescued by simultaneous ablation of PTP1B in these neurons (Qin et al., 2020).In the ketamine model, sensorimotor gating deficits and spatial memory deficits were also partly correlated to defective endocannabinoid mobilization in response to the trkB receptor agonist 7,8-dihydroxyflavone (Qin et al., 2021).Glutamatergic neuron-specific ablation of PTP1B prevented ketamine’s effect to reduce trkB-dependent endocannabinoid mobilization from layer 2/3 pyramidal neurons (Figure 1B). In light of the report showing that presynaptic NMDA receptors regulate brain-derived neurotrophic factor release to modulate post-synaptic trkB signaling (Park et al., 2014), it will be important to determine how PTP1B at the presynaptic site affects this process in the context of schizophrenia (and other neurological disorders).
Together, these recent studies reveal novel aspects of NMDA receptor signaling affected by PTP1B in two preclinical models of neurological disorders:Alzheimer’s disease and schizophrenia.In the hAPP-J20 model of Alzheimer’s disease, PTP1B hyperactivity impairs presynaptic NMDA receptor-dependent LTP at the CA3:CA1 hippocampal synapse(Figure 1A). In a model of schizophrenia,the NMDA receptor antagonist ketamine causes dis-inhibition of PTP1B that impairs trkB-dependent endocannabinoid mobilization in post-synaptic neurons of the medial prefrontal cortex (Figure 1B).Importantly, these studies also show the therapeutic potential of PTP1B inhibitors to treat these disorders.
This work was supported by grants from the Heart and Stroke Foundation of Canada (G-13-0002596 & G-18-0022157,to HHC; G-16-00014085, to AFRS), Ontario Mental Health Foundation (to HHC), the Canadian Institutes of Health Research(201610PJT #376403, to HHC; 201610PJT#376503, to AFRS), the Natural Science and Engineering Research Council of Canada (RGPIN/06212-2014, to HHC;RGPIN/2016-04985, to AFRS). HHC is also supported by a Mid-Career Investigator Award (grant # 7506) from the Heart and Stroke Foundation of Ontario.
Alexandre F. R. Stewart*,
Hsiao-Huei Chen*Department of Biochemistry, Microbiology and Immunology, University of Ottawa, and University of Ottawa Heart Institute, Ottawa, ON, Canada(Stewart AFR)Centre for Infection, Immunity and Inflammation,Ottawa, ON, Canada (Stewart AFR, Chen HH)Medicine, Cellular and Molecular Medicine,University of Ottawa Brain and Mind Institute,Ottawa Hospital Research Institute, Ottawa, ON,Canada (Chen HH)
*Correspondence to: Alexandre F. R. Stewart, PhD,astewart@ottawaheart.ca; Hsiao-Huei Chen, PhD,hchen@uottawa.ca.https://orcid.org/0000-0003-2673-9164(Alexandre F. R. Stewart);https://orcid.org/0000-0003-2914-6057(Hsiao-Huei Chen)
Date of submission: July 24, 2021
Date of decision: September 27, 2021
Date of acceptance: October 28, 2021
Date of web publication: February 28, 2022
https://doi.org/10.4103/1673-5374.335793
How to cite this article:Stewart AFR, Chen HH(2022) N-methyl-D-aspartate receptor functionsaltered by neuronal PTP1B activation in Alzheimer’s disease and schizophrenia models. Neural RegenRes 17(10):2208-2210.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Functional in vivo assessment of stem cell-secreted prooligodendroglial factors
- iGluR expression in the hippocampal formation, entorhinal cortex,and superior temporal gyrus in Alzheimer’s disease
- Exploiting Caenorhabditis elegans to discover human gut microbiotamediated intervention strategies in protein conformational diseases
- Aminopeptidase A and dipeptidyl peptidase 4: a pathogenic duo in Alzheimer’s disease?
- Ubiquitin homeostasis disruption,a common cause of proteostasis collapse in amyotrophic lateral sclerosis?
- Novel insights into the pathogenesis of tendon injury:mechanotransduction and neuroplasticity