
Expression and regulatory network of long noncoding RNA in rats after spinal cord hemisection injury
2022-03-09 07:22WeiLiuJinChengTaoShengZeZhuChaoLunDaiYaXianWangBinYuChunYaoYuYuSun
中国神经再生研究(英文版) 2022年10期
Wei Liu, Jin-Cheng Tao, Sheng-Ze Zhu, Chao-Lun Dai, Ya-Xian Wang, Bin Yu,Chun Yao,*, Yu-Yu Sun
Abstract Long noncoding RNAs (lncRNAs) participate in a variety of biological processes and diseases. However, the expression and function of lncRNAs after spinal cord injury has not been extensively analyzed. In this study of right side hemisection of the spinal cord at T10, we detected the expression of lncRNAs in the proximal tissue of T10 lamina at different time points and found 445 lncRNAs and 6522 mRNA were differentially expressed. We divided the differentially expressed lncRNAs into 26 expression trends and analyzed Profile 25 and Profile 2, the two expression trends with the most significant difference. Our results showed that the expression of 68 lncRNAs in Profile 25 rose first and remained high 3 days post-injury. There were 387 mRNAs co-expressed with the 68 lncRNAs in Profile 25. The co-expression network showed that the co-expressed genes were mainly enriched in cell division, inflammatory response, FcγR-mediated cell phagocytosis signaling pathway, cell cycle and apoptosis. The expression of 56 lncRNAs in Profile2 first declined and remained low after 3 days post-injury.There were 387 mRNAs co-expressed with the 56 lncRNAs in Profile 2. The co-expression network showed that the co-expressed genes were mainly enriched in the chemical synaptic transmission process and in the signaling pathway of neuroactive ligand-receptor interaction. The results provided the expression and regulatory network of the main lncRNAs after spinal cord injury and clarified their co-expressed gene enriched biological processes and signaling pathways.These findings provide a new direction for the clinical treatment of spinal cord injury.
Key Words: bioinformatic analysis; biological process; gene ontology analysis; inflammatory response; Kyoto encyclopedia of genes and genomes analysis; long noncoding RNAs; regulatory network; RNA sequencing; spinal cord injury; synaptic transmission
Introduction
Injury to the central nervous system includes brain injury and spinal cord injury (SCI), which often results in losing part or all of the sensory and motor functions distal to the injury site. SCI causes great pain to patients and thetime and money spent on medical care bring a heavy burden to the family of patients (Lin, 2019). In the past, it was generally thought that the central nervous system of adult mammals could not regenerate. The main reasons are that the neurons in the central nervous system have lose the ability to regeneration at maturity (Kwon et al., 2016) and that the microenvironment of the central nervous system after injury is not conducive to nerve regeneration (Tedeschi and Bradke, 2017; Yang et al., 2021; Bai et al., 2021).SCI is identified as the injury caused by direct or indirect external factors.There are various clinical and pathological changes after SCI, both motor and sensory, including sphincter dysfunction and abnormal muscle tension. After the physical injury, a series of physiological and pathological responses follow:(i) Local ischemia, edema and other severe inflammatory responses resulting from vascular rupture (Mortazavi et al., 2015), (ii) Neuronal degeneration,apoptosis and autophagy, (iii) Nerve regeneration controlled by astrocytes that promote it in the early stage and inhibit it in the late stage and (iv)Inflammation aggravated by macrophage activation (Fitch and Silver, 2008).The combined effect of the above physiological and pathological responses leads to an unfavorable microenvironment for nerve regeneration.
In the human genome, about 50% of DNA can be transcribed into RNA.However, mRNA accounts for only 1.2% of the transcribed RNA, and the remaining RNA, termed noncoding RNA, cannot be translated into proteins(Jarroux et al., 2017). Among them, a class of noncoding RNAs with a length of more than 200 nucleotides (nt) are termed long noncoding RNAs (lncRNAs)(Wilusz et al., 2009; Zhang et al., 2019). A large number of studies have explored mRNA levels during the functional repair after SCI, in particular the phosphatase and tensin homolog deleted on chromosome ten (PTEN),which defies the traditional view that the central nervous system cannot be repaired (Ohtake et al., 2014). However, there have been few studies on the expression and role of lncRNAs after SCI. The co-expression of lncRNAs with mRNAs suggests the importance of understanding the role of lncRNAs when developing new therapies to regulate the physiological and pathological responses after injury and to improve rehabilitation.
This study selected the hemisection SCI model with its high repeatability(Kozuka et al., 2016). In the 7 days following injury, we compared the changes of lncRNAs and mRNAs in the sham group with those of the SCI group to identify differentially expressed lncRNAs and mRNAs. The results would clarify the enriched biological processes involving lncRNA and their co-expressed genes after the hemisection SCI.
Materials and Methods
Animal surgery
The animal experiments were ethically approved by the Administration Committee of Experimental Animals, Jiangsu Province, China (approval No.20180304-008) on March 4, 2018. For the convenience of animal care, female Sprague-Dawley rats (aged 2 months old; weight 200-220 g), approved by the Administration Committee of Experimental Animals, Jiangsu Province, China(license Nos. SCXK (Su) 2019-0001 and SYXK (Su) 2017-0046), were used in this study. The rats were raised in the specific-pathogen-free conditions, with only five rats in each cage (temperature 22°C, humidity 50-65%, 12/12-hour light/dark and had free access to food and water). All animal experiments were conducted according to the nursing guidelines of Experimental Animal Center of Nantong University and were designed and reported according to the Animal Research: Reporting ofIn VivoExperiments (ARRIVE) guidelines(Percie du Sert et al., 2020).
In this study, rats were allocated to the two groups using the random principle. In the sham group (n= 72) T10 was only exposed. In the SCI group(n= 72), the spinal cord was exposed after resection of T10 lamina with the right hemisection. During the operation, rats were given an intraperitoneal injection of compound anesthetics (85 mg/kg chloral hydrates, 42 mg/kg magnesium sulphates and 17 mg/kg pentobarbital sodium: MilliporeSigma,Burlington, MA, USA and Merck KGaA, Darmstadt, Germany). When the rats were completely anesthetized, they were placed on a 37°C thermostatic electric blanket. Christensen’s surgical method was used (Christensen et al.,1996) where the skin, fascia, and muscle were longitudinally cut at T9-T10,and the T10 lamina was removed to expose the spinal cord to perform a right hemisection using an ophthalmic iris knife (Beaver-Visitec International,Waltham, MA, USA; Wu et al., 2019). After the operation, the muscles, fascia and skin were sutured with absorbable sutures. When the rats were fully awake, they were moved to a clean and dry cage and provided with sufficient water and organic feed.
RNA-sequencing and bioinformatic analysis
The rats selected at each time point after injury, days 0 (immediately after the injury), 1, 3, 7, were given an intraperitoneal injection of compound anesthetics during the operation to collect 5 mm of spinal cord tissue,proximal to the injury. When the operation was complete, each rat was sacrificed by an injection of excess compound anesthetic. To achieve three repeated samples at each time point, a sample was taken from each of six rats to reduce the impact of individual differences. The total RNA from spinal cord tissue was extracted by the mirVana miRNA isolation kit (Ambion,Austin, TX, USA) and sequenced by Shanghai OE Biotechnology Co., Ltd.(Shanghai, China). The RNA library was sequenced on the Illumina HiSeq X10 platform (Illumina, Santiago, CA, USA) and produced 150-bp pair-end reads. First, the NGS QC Toolkit (http://www.nipgr.res.in/ngsqctoolkit.html)was used to process the raw data (the original reads) of FASTO format, and RPM (reads, mapped by splicing, per million) was taken as the expression of lncRNA and mRNA. Differential expression analysis was conducted using DESeq R package (www.Bioconductor.org) (Anders and Huber, 2010). LncRNAs with fold change > 2 andP-value ≤ 0.05 were considered as differentially expressed. The differentially expressed lncRNAs and mRNAs that overlapped between the sham and the SCI groups were removed, and the remainder was identified as differentially expressed lncRNAs and mRNAs after SCI. TheP-value was then calculated for each lncRNA and mRNA (negative binomial distribution test), and theP-value was corrected for multiple hypothesis tests using the false discovery rate error control method to obtain aQ-value.Unsupervised hierarchical clustering was performed on differently expressed lncRNA and mRNA, then the expression patterns of differentially expressed genes in different samples were displayed in the form of heat maps. The STEM software (http://www.sb.cs.cmu.edu/stem/) was used to describe the expression trend diagram for time series analysis of different sample materials.
Gene ontology (GO) (The Gene Ontology Consortium, 2019) and KEGG(Kyoto Encyclopedia of Genes and Genomes) analyses were conducted,according to the previous method (Mi et al., 2017). The GO were obtained from the mapped translated protein sequences and homology searches against Swissprot using BLASTX in DIAMOND v0.9.30 (University of Tübingen,Tübingen, Germany) with an E-value cut-off of 1 × e-5 to retrieve the identifier(ID) mapping. The KEGG annotation was obtained by submitting the coding sequences of mapped genes onto the KASS-KEGG Automatic Annotation Server (Kyoto University, Kyoto, Japan). Enrichment analysis was performed via a hypergeometric test and statistical significance was considered atP≤ 0.05(Shanghai OE Biotechnology Co., Ltd.).
Real-time polymerase chain reaction
We collected total RNA from the proximal 5 mm spinal cord tissues after SCI with TRIzol reagent (Gibco, Grand Island, NY, USA). HiScript III RT SuperMix 100 for qPCR (+gDNA wiper) (Vazyme, Nanjing, China) was used to reverse transcript RNA into complementary DNA. Real-time polymerase chain reaction was performed with ChamQ SYBR qPCR Master Mix (Vazyme) on a StepOne Real-time PCR System (Applied Biosystems, Foster City, CA, USA). Primer sequences were listed in Additional Table 1. The results were calculated by 2-ΔCT.
Statistical analysis
No statistical methods were used to predetermine sample sizes; however,our sample sizes were similar to those reported in a previous publication (Wu et al., 2019). There was no loss of experimental animals. All evaluators were blinded to the animal groups. Data processing and analysis were performed using GraphPad 8 (GraphPad Software, Inc., La Jolla, CA, USA, www. graphpad.com) and SPSS 20.0 (IBM, Armonk, NY, USA) statistical analysis software. We used Student’st-test for RT-PCR to analyze the difference between groups with double tail test. One-way analysis of variance was performed for the sham and the SCI groups respectively to exclude the differentially expressed lncRNAs and mRNAs which overlapped both the sham and SCI groups. The mean and standard error (SEM) were then calculated.P< 0.05 was considered statistically significant.
Results
Differentially expressed lncRNAs in proximal spinal cord after SCI
We used a rat T10 hemisection injury model to detect the lncRNA changes after SCI. RNA-sequencing analysis was carried out on the proximal hemisection spinal cord tissue at different time points after SCI. Depending on the standard ofQ-value less than 0.01, there were 445 differentially expressed lncRNAs between the sham and SCI groups (Figure 1A and Additional Table 2)and 6522 differentially expressed mRNAs (Figure 1B and Additional Table 3)after spinal cord hemisection injury. In the heat map of lncRNAs and mRNAs,we found that the expression trend of lncRNAs and mRNAs at day 0 were similar to that of day 1, while those at days 3 and 7 were similar.
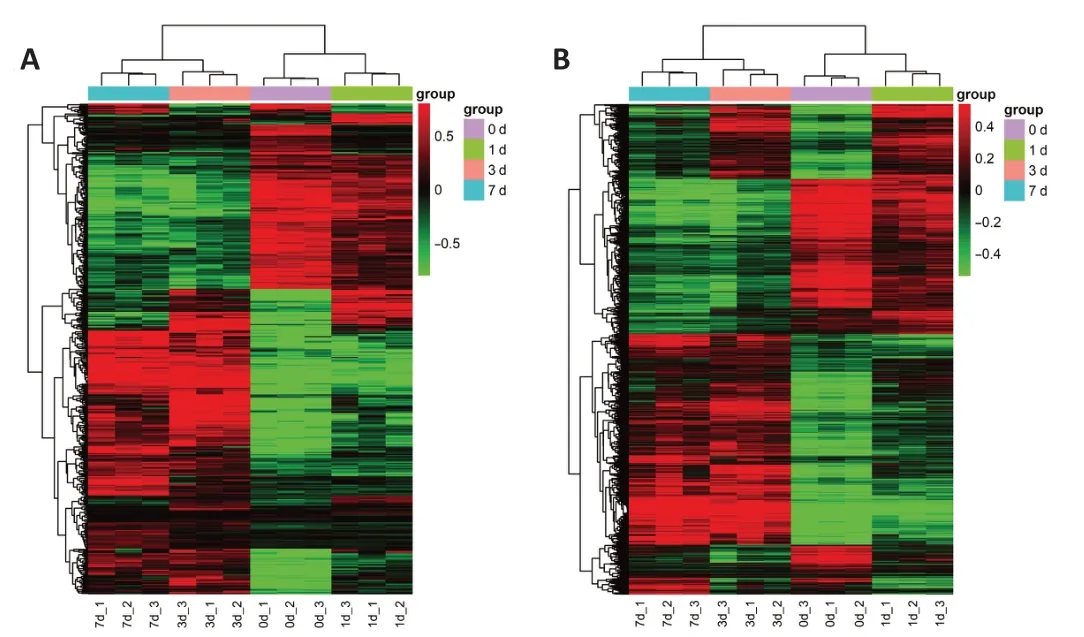
Figure 1 |Bioinformatic analysis of dysregulated lncRNA and mRNA after spinal cord injury.(A) Heatmap of dysregulated lncRNAs at 0, 1, 3, and 7 days after spinal cord injury. The X-axis is the sample name. The Y-axis is the name of lncRNAs, and their detailed names are in the Additional Table 2. (B) Heatmap of dysregulated mRNAs at 0, 1, 3, and 7 days after spinal cord injury. The X-axis is the sample name. The Y-axis is the name of lncRNAs,and their detailed names are in the Additional Table 3. The expression trends of lncRNAs and mRNAs on day 0 were similar to that on day 1, whereas those on day 3 were similar to day 7. Red indicates up-regulated RNAs while green indicates down-regulated RNAs. 0 d_1: The sample 1 of 0 day; 0 d_2: the sample 2 of 0 day; 0 d_3: the sample 3 of 0 day; 1 d_1: the sample 1 of 1 day; 1 d_2: the sample 2 of 1 day; 1 d_3: the sample 3 of 1 day; 3 d_1: the sample 1 of 3 days; 3 d_2: the sample 2 of 3 days; 3 d_3: the sample 3 of 3 days;7 d_1: the sample 1 of 7 days; 7 d_2: the sample 2 of 7 days; 7 d_3: the sample 3 of 7 days; LncRNA: long noncoding RNA; mRNA: messenger RNA.
As shown in Figure 2, we divided the differentially expressed lncRNAs into 26 expression trends. Among them, eight trend profiles had significantP-value,shown in red. We then picked two tendencies with the most significant difference, Profile 25 and Profile 2, for validation. Interestingly, expressions of lncRNAs in Profile 25 rose at first and then plateaued while expressions of lncRNAs in Profile 2 declined over the first three days and then remained constant.
Functional analysis of differentially expressed lncRNAs in Profile 25
After SCI the expressions of the lncRNAs in Profile 25 rose first and then remained high after 3 days. There were 68 lncRNAs in Profile 25 (Figure 3A). We then randomly selected NONRATT001559.2, NONRATT005821.2,NONRATT027850.2 and NONRATT020851.2 for RT-PCR validation. The RT-PCR results of the selected lncRNAs were very consistent with the sequencing data(Figure 3B).
There were 387 mRNAs co-expressed with the 68 lncRNAs in Profile 25. A coexpression network was constructed for the differentially expressed lncRNAs and co-expressed mRNAs in Profile 25 (Figure 3C) to show the interactions between lncRNAs and mRNAs (Additional Table 4).
GO term enrichments were performed for the co-expressed mRNAs in Profile 25. The top 20 GO terms are shown, which suggest that the co-expressed genes were mainly enriched in processes of cell division and inflammatory response (Figure 3D and Additional Table 5). KEGG signaling pathway enrichment analysis was also conducted for the co-expressed mRNAs in Profile 25. The top 20 pathways are shown, which suggest that the coexpressed genes were mainly enriched in the FcγR-mediated cell phagocytosis signaling pathway, cell cycle and apoptosis (Figure 3E and Additional Table 6).The functional analysis of co-expressed genes in Profile 25 indicated that the lncRNAs in this profile were involved in cell cycle, apoptosis and inflammation.

Figure 2 |Bioinformatic analysis of the expression trend of differentially expressed lncRNA after SCI.Trends with significant difference (P < 0.05) were marked in red. 0 to 3 indicate days 0,1, 3 and 7, respectively. The vertical axis indicates the overall trend of all lncRNAs in a module. LncRNA: Long noncoding RNA; SCI: spinal cord injury.
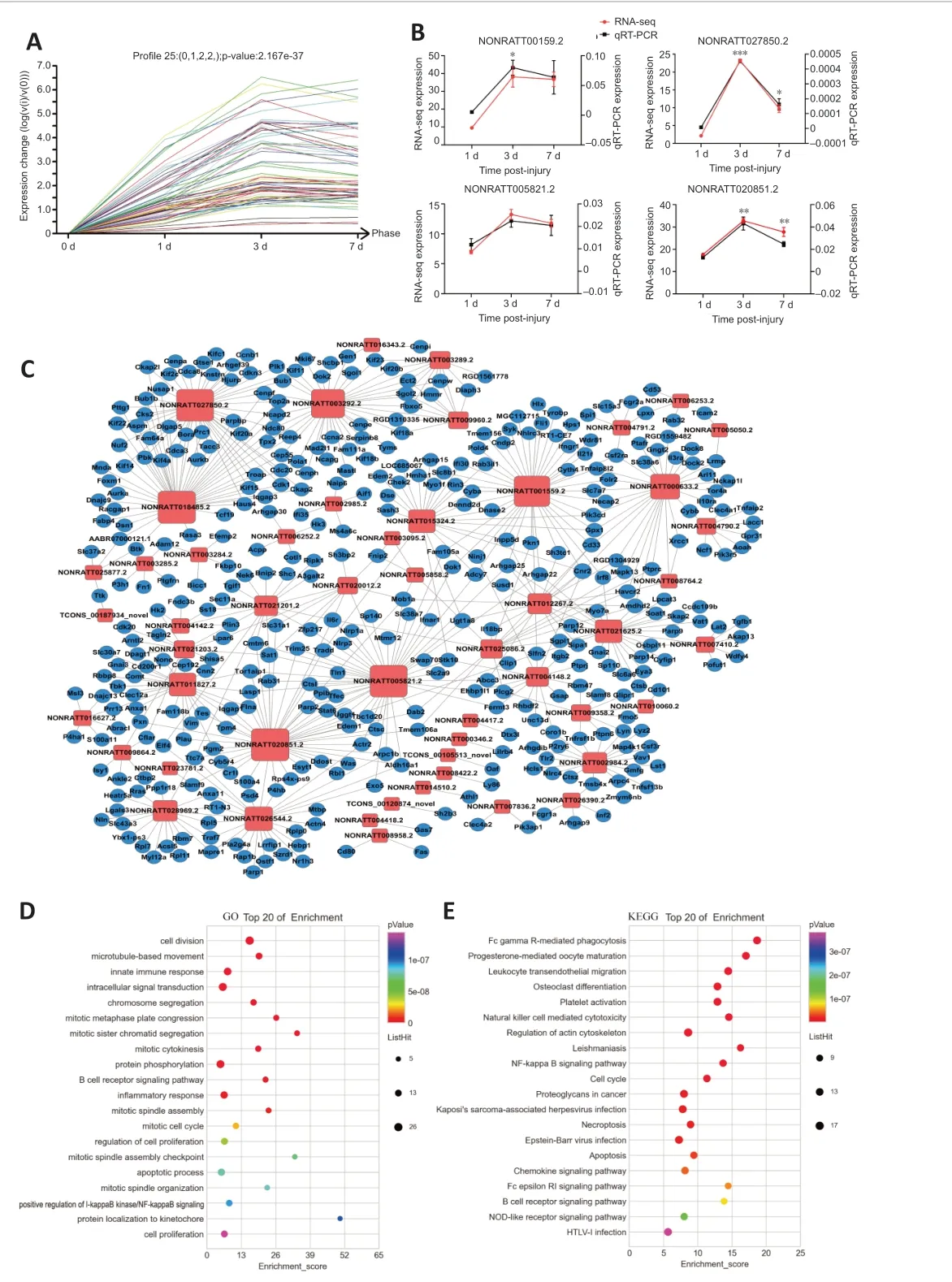
Figure 3 |RT-PCR validation and bioinformatic analysis of lncRNAs after SCI in Profile 25.(A) RNA-sequencing trend diagram of lncRNAs in Profile 25. (B) RT-PCR validation of randomly selected lncRNAs in Profile 25. Data are expressed as mean ± SEM (n = 3 for each group). *P < 0.05, **P < 0.01, ***P < 0.001, vs. 1 day (Student’s t-test). (C) Coexpression network for the differentially expressed lncRNA and co-expressed mRNA in Profile 25 after SCI. The blue round nodes represent mRNA, and the red square nodes indicate lncRNA. The more co-expressed genes of the lncRNA, the larger the size of the red box. Solid line between nodes indicated a positively correlated relationship between lncRNA and mRNA. (D) Bubble diagram of the top 20 enriched GO terms of coexpressed mRNAs in Profile 25. The y-axis shows the enriched top 20 GO terms, and the x-axis represents the enrichment score. The size of the bubble represents the number of co-expressed genes in the GO term. The color of the bubble represents the P-value of enriched GO term. (E) Bubble diagram of the top 20 enriched KEGG pathways of coexpressed mRNAs in Profile 25. The y-axis shows the enriched top 20 pathways, and the x-axis represents their enrichment score. The size of the bubble gives a measure of the number of co-expressed genes in the pathway. The color of the bubble represents the P-value of the enriched pathway. GO: Gene ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; LncRNA: long noncoding RNA; mRNA: messenger RNA; RT-PCR: real-time polymerase chain reaction; SCI: Spinal cord injury. The full name of mRNA can be found in the Additional Table 3.
Functional analysis of differential expression lncRNAs in Profile 2
The expressions of lncRNAs in Profile 2 declined first and then remained low after 3 days post-injury. There were 56 lncRNAs in Profile 2 (Figure 4A). We then randomly selected NONRATT002333.2, NONRATT009996.2,NONRATT026122.2 and TCONS_00083425_novel for RT-PCR validation. The RT-PCR results of selected lncRNAs were very consistent with the sequencing data (Figure 4B).
There were 387 mRNAs co-expressed with the 56 lncRNAs in Profile 2. A coexpression network was constructed for the differentially expressed lncRNA and co-expressed mRNA in Profile 2 (Figure 4C) to show the interactions between lncRNAs and mRNAs (Additional Table 7).
GO term enrichments were performed for the co-expressed mRNAs in Profile 2. The top 20 GO terms are shown, which suggested that the co-expressed genes were mainly enriched in the chemical synaptic transmission process(Figure 4D and Additional Table 8). KEGG signaling pathway enrichment analysis was conducted for the co-expressed mRNAs in Profile 2. The top 20 pathways are shown, which suggest that the co-expressed genes were mainly enriched in the signaling pathway of neuroactive ligand-receptor interaction(Figure 4E and Additional Table 9).
Discussion
SCI continues to be a difficult global clinical problem. Rehabilitation and treatments after injury often bring a heavy economic burden on patients and their families. In recent years, many researchers have devoted much effort to improve regeneration and rehabilitation after SCI, but with limited effect.In this study, we analyzed the whole transcriptome of the injured spinal cord and identified dysregulated lncRNAs after SCI.
SCI is divided into primary injury and secondary injury. Primary injury is often caused by sudden spinal trauma. Subsequently, primary injury induces secondary injury, causing further chemical and mechanical injury to spinal cord tissue (Orr and Gensel, 2018). Secondary injury can be divided into three stages: acute (0-48 hours), sub-acute (first 2 weeks) and chronic injury(from days to years) (Anjum et al., 2020; Zhu et al., 2021). The acute injury stage mainly includes vascular damage, ionic imbalance, excitotoxicity, free radical production, increased calcium influx, lipid peroxidation, inflammation,oedema and necrosis (Phang et al., 2015; Alizadeh et al., 2019). The sub-acute stage includes neuronal apoptosis, axon demyelination, Waller degeneration,axon remodeling and glial scar formation (Alizadeh et al., 2019). The chronic injury stage includes cyst formation, axon necrosis and glial scar maturation(Kjell and Olson, 2016; Tran et al., 2018). In this study, we focused on the acute injury stage and the sub-acute injury stage, expecting to find key lncRNAs that participate in post-injury processes at these early stages after SCI.

Figure 4 |RT-PCR validation and bioinformatic analysis of lncRNAs in Profile 2.(A) RNA-sequencing trend diagram of lncRNAs in Profile 2. (B) RT-PCR validation of randomly selected lncRNAs in Profile 2. Data are expressed as mean ± SEM (n = 3 for each group). *P < 0.05, vs. 1 day (Student’s t-test). (C) The co-expression network for the differentially expressed lncRNA and co-expressed mRNA in Profile 2 after SCI. The blue round nodes represent mRNA, and the red square nodes indicate lncRNA. The more coexpressed genes of the lncRNA, the larger the size of the red box. The solid line between nodes indicated the positively correlated relationship between lncRNA and mRNA. (D)Bubble diagram of the top 20 enriched GO terms of co-expressed mRNAs in Profile 2. The y-axis shows the enriched top 20 GO terms, and the x-axis represents their enrichment score. The size of the bubble represents the number of co-expressed genes in the GO term. The color of the bubble represents the P-value of enriched GO term. (E) Bubble diagram of the top 20 enriched KEGG pathways of co-expressed mRNAs in Profile 2. The y-axis shows the enriched top 20 pathways, and the x-axis represents their enrichment score. The size of the bubble represents the number of co-expressed genes in the pathway. The color of the bubble represents the P-value of enriched pathway. GO: Gene ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; LncRNA: long noncoding RNA; mRNA: messenger RNA; RT-PCR: real-time polymerase chain reaction; SCI: Spinal cord injury. The full name of mRNA can be found in the Additional Table 3.
The differentially co-expressed genes of lncRNAs in Profile 25 (which were upregulated after SCI) were enriched in cell mitosis, cell proliferation, apoptosis and inflammatory response. Astrocytes and microglia play a key role during these processes. The role of astrocytes has been shown to regulate neutrophils, improve the resistance to bacterial infection and prevent bacteria from crossing the blood-brain barrier (Xie et al., 2010). Astrocytes could secrete a large number of pro-inflammatory and anti-inflammatory factors (Sofroniew, 2015). Others revealed that astrocytes can induce the differentiation and proliferation of microglia and promote the phagocytosis of microglia (Zeinstra et al., 2000). The importance of astrocytes and microglia in the early stage of SCI indicated the necessity to explore the cellular and molecular changes in astrocytes and microglia (Figure 5).The differentially co-expressed genes of lncRNAs in Profile 2 (which were down-regulated after SCI) were enriched in synaptic transmission, ion transmembrane transport, neuronal excitation, and movement. In the central nervous system, the information transmission between neurons mainly depends on synaptic transmission (Jackman and Regehr, 2017). Studies have shown that astrocytes, microglia and oligodendrocytes participate in synaptic plasticity regulation (Bar and Barak, 2019; Durkee and Araque, 2019).The major roles of astrocytes in synapses are neurotransmitter clearance,neurotransmitter release and regulation of ion balance (Goubard et al.,2011; Perez-Alvarez et al., 2014; Murphy-Royal et al., 2017). Microglia also play a very important role in synapses by regulating the release of cytokines and neurotrophic factors and in synaptic pruning (Vezzani and Viviani,2015; Hammond et al., 2018). The oligodendrocytes form new myelin or change myelin characteristics in synapses (Sancho et al., 2021). They receive glutamatergic synapses from neurons to detect neuronal activity (Bergles et al., 2000). The pre-myelinating oligodendrocytes can activate Nav1.2 (voltagegated channel alpha subunit 2)-driven depolarizations, in order to respond to the glutamatergic input from neighboring neurons (Berret et al., 2017). In addition, the balance between axon regeneration and synaptic function is also important for the reconstruction of functional neuronal circuits after nerve injury (Kiyoshi and Tedeschi, 2020) (Figure 5).
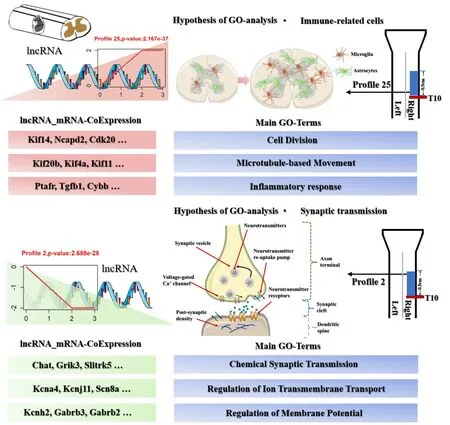
Figure 5 |Schematic diagram of changed lncRNAs, enriched biological processes,and key mRNAs in Profiles 25 and 2 after spinal cord injury.Cdk20: Cyclin-dependent kinase 20; Chat: choline O-acetyltransferase; Cybb: cytochrome b-245, beta polypeptide; Gabrb2: gamma-aminobutyric acid A receptor, beta 2; Gabrb3:gamma-aminobutyric acid A receptor, beta 3; GO: Gene ontology; Grik3: glutamate receptor, ionotropic, kainate 3; Kcna4: potassium channel, voltage gated shaker related subfamily A, member 4; Kcnh2: potassium channel, voltage gated Eag related subfamily H, member 2; Kcnj11: potassium channel, inwardly rectifying subfamily J, member 11;Kif11: kinesin family member 11; Kif14: kinesin family member 14; Kif20b: kinesin family member 20B; Kif4a: kinesin family member 4A; LncRNA: long noncoding RNA; mRNA:messenger RNA; Ncapd2: non-SMC condensin I complex, subunit D2; Ptafr: plateletactivating factor receptor; SCI: spinal cord injury; Scn8a: sodium channel, voltage gated,type VIII, alpha subunit; Slitrk5: SLIT and NTRK-like family, member 5; Tgヅ1: transforming growth factor beta 1.
Common spinal cord injury models include contusion, full transection and hemisection injury. Contusions can better simulate clinical spinal cord injuries from car accidents, but as with all mechanical models, it is difficult to precisely control the biomechanics to produce consistent and repeatable spinal cord injuries. The full transection injury model ensures that natural or post-intervention recovery is due to repair after SCI (Jones et al., 2012), but this model is not ideal for exploring the complex pathophysiology involved in spinal cord injury because transection is not common in clinical settings.The hemisection injury model simulates an injury that is more readily seen clinically and provides a comparison between damaged and healthy fibers in the same animal. For example, hemictomies can be used to examine motor function and recovery of different spinal tracts, or to compare functional changes in contralateral and ipsilateral lesions (Alilain et al., 2011). Therefore,in this study, we selected the semi-transverse damage model with good repeatability and stability. In the future, we will also use a variety of models to further explore the dysregulated lncRNAs after SCI.
In conclusion, we have identified a set of dysregulated lncRNAs during the early period after SCI and obtained the enriched biological processes and signaling pathways of the co-expressed genes. Our research has provided a potential research direction for SCI treatment. However, because the RNAseq samples were obtained from mixed tissues, it was hard to determine which differentially expressed genes were associated with what cell type.And the research of the mechanism is not deep enough because we only conducted simple expression verification without any functional experiments.In future, we should perform single-cell RNA-seq to further address this issue and conduct in-depth mechanism research. The next focus would be on the chronic phase after SCI to obtain a more comprehensive view of the dysregulated lncRNAs after SCI and to identify some cells with significant changes in lncRNAs.
Author contributions:Study conception and design: CY, YYS; experiment implementation and data analysis: WL, JCT, SZZ, CLD, YXW, BY; reagents/materials/analysis support: CY, YYS; manuscript writing: WL, CY. All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare that they have no conflicts of interest.
Availability of data and materials:All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under theidentical terms.
Additional files:
Additional Table 1: The primer sequences of long noncoding RNAs for quantitative real-time polymerase chain reaction validation.
Additional Table 2: The differentially expressed long noncoding RNAs at different time points (0, 1, 3, 7 days) after spinal cord injury.
Additional Table 3: The differentially expressed mRNAs at different time points (0, 1, 3, 7 days) after spinal cord injury.
Additional Table 4: The data of co-expression network between long
noncoding RNAs and co-expressed genes in Profile 25.
Additional Table 5: The enriched top 20 GO terms of co-expressed genes in Profile 25.
Additional Table 6: The enriched top 20 KEGG pathways of co-expressed genes in Profile 25.
Additional Table 7: The data of co-expression network between long noncoding RNAs and co-expressed genes in Profile 2.
Additional Table 8: The enriched top 20 GO terms of co-expressed genes in Profile 2.
Additional Table 9: The enriched top 20 KEGG pathways of co-expressed genes in Profile 2.

- 中国神经再生研究(英文版)的其它文章
- Functional in vivo assessment of stem cell-secreted prooligodendroglial factors
- iGluR expression in the hippocampal formation, entorhinal cortex,and superior temporal gyrus in Alzheimer’s disease
- Exploiting Caenorhabditis elegans to discover human gut microbiotamediated intervention strategies in protein conformational diseases
- N-methyl-D-aspartate receptor functions altered by neuronal PTP1B activation in Alzheimer’s disease and schizophrenia models
- Aminopeptidase A and dipeptidyl peptidase 4: a pathogenic duo in Alzheimer’s disease?
- Ubiquitin homeostasis disruption,a common cause of proteostasis collapse in amyotrophic lateral sclerosis?