
A neurovascular unit-on-a-chip: culture and differentiation of human neural stem cells in a three-dimensional microfluidic environment
2022-03-09 07:31WenJuanWeiYaChenWangXinGuanWeiGongChenJingLiu
中国神经再生研究(英文版) 2022年10期
Wen-Juan Wei, Ya-Chen Wang, Xin Guan, Wei-Gong Chen, Jing Liu,*
Abstract Biological studies typically rely on a simple monolayer cell culture, which does not reflect the complex functional characteristics of human tissues and organs,or their real response to external stimuli. Microfluidic technology has advantages of high-throughput screening, accurate control of the fluid velocity, low cell consumption, long-term culture, and high integration. By combining the multipotential differentiation of neural stem cells with high throughput and the integrated characteristics of microfluidic technology, an in vitro model of a functionalized neurovascular unit was established using human neural stem cellderived neurons, astrocytes, oligodendrocytes, and a functional microvascular barrier. The model comprises a multi-layer vertical neural module and vascular module, both of which were connected with a syringe pump. This provides controllable conditions for cell inoculation and nutrient supply, and simultaneously simulates the process of ischemic/hypoxic injury and the process of inflammatory factors in the circulatory system passing through the blood-brain barrier and then acting on the nerve tissue in the brain. The in vitro functionalized neurovascular unit model will be conducive to central nervous system disease research,drug screening, and new drug development.
Key Words: (neural) differentiation; astrocyte; blood-brain barrier; brain microvascular endothelial cells; central nervous system; microfluidics; neural stem cells; neuron; neurovascular unit; oligodendrocyte; organ-on-a-chip
Introduction
In vitromodels are the most valuable tools for studying cell behavior in a controlled and replicable environment. However, most biological studies over the past century relied on a simple monolayer cell culture. These simplified methods do not reflect the complex functional characteristics of humantissues and organs, or their real response to external stimuli. Although animal experiments can provide somein vivoinformation, they still have limitations,such as high cost, poor repeatability, differences between species, and insufficient ability to predict the real human response.
Researchers first reported a microfluidic device to construct cell modules and simulate the microenvironment in 2004 (Andersson and van den Berg, 2004;Sin et al., 2004). The microfluidic device, as a bioreactor, was called “organon-a-chip”. After more than 10 years of development, these organ-on-a-chips have successfully been used to reproduce many tissues and organs (Oleaga et al., 2016; Wu et al., 2020; Rothbauer et al., 2021) such as lung (Stucki et al., 2015; Shrestha et al., 2020), liver (Banaeiyan et al., 2017; Moradi et al.,2020), kidney (Zhou et al., 2016; Lee and Kim, 2018), and skin (Wufuer et al., 2016; Lee et al., 2017). These precisely constructed models are used to clarify physiological phenomena that are difficult to dynamically observe,systematically evaluate, and quantifyin vivo, and can serve as effective tools for high-throughput screening of therapeutic drugs. Microfluidic technology has advantages of high-throughput screening, accurate control of the fluid velocity, low cell consumption, long-term culture, and high integration(Sackmann et al., 2014; van Duinen et al., 2015). Through microfluidic ortissue engineering technology, different cells of the same organ are integrated into a limited culture space according to a specified organization to form a living cell structure unit with a certain tissue structure and physiological function. Reardon (2015) reported that the organ-on-a-chip is a revolutionary technology that may replace animal experiments.
The brain, as part of the central nervous system (CNS) of the human body,is an important target organ for drug screening and toxicity testing. The concept of the neurovascular unit (NVU) was first proposed by Harder et al.(2002). NVU comprises endothelial cells, extracellular matrix, astrocytes,pericytes, neurons and their axons, and other supporting cells (microglia,oligodendrocytes). The components are linked reciprocally and intimately,generating an anatomically and functionally efficient system that regulates the cerebral blood flow (McConnell et al., 2017; De Luca et al., 2020; Caffrey et al.,2021; Liu et al., 2021; Ye et al., 2021). NVU is the basic unit of the structure and function of the CNS. The NVU system is different from traditional bloodbrain barrier (BBB) models that do not include glia or neurons with the other CNS elements. In the last 20 years, physiologic researchers have verified that cells in the vasculature intercellularly communicated with adjoining glia and neurons strongly. Thus, instead of working independently, BBB functions as a module in a multicellular NVU with astrocytes, neurons, oligodendroglia,microglia, and blood vessels (Muoio et al., 2014; McConnell et al., 2017;Bhalerao et al., 2020). As a special structure in the brain, the NVU has evident regulatory effects on the blood flow via the vasculature. Because the NVU is an important structure for maintaining brain homeostasis, the destruction or dysfunction of any component may lead to CNS disease (Sweeney et al., 2016;Wareham and Calkins, 2020; Yu et al., 2020; Holloway et al., 2021).
Neural stem cells (NSCs), as pluripotent cells in the CNS, have high selfrenewal capacity. NSCs are of great significance to embryonic development,neurogenesis, and tissue homeostasis. In addition, they are suitable cellular systems for regenerative medicine and developmental biologyin vitroresearch because of the potential to differentiate into almost all neuron lineages (Obernier and Alvarez-Buylla, 2019).
We aim to combine the benefits ofin vitromicrofluidic technologies with the neurogenic differentiation potential of stem cells. In this study, we constructed a functionalized NVU model based on a microfluidic device using primary human NSCs (hNSCs) and brain microvascular endothelial cells(BMECs) under dynamic flow conditions. The construction of the NVU-ona-chip will be conducive to CNS disease research, drug screening, and new drug development. This model will become a valuable tool for studying cell behavior in a controllable and reproducible environmentin vitro.
Materials and Methods
Cell isolation, culture, and differentiation
The aborted embryos were collected at 6-12 weeks of pregnancy, and hNSCs were isolated and culturedin vitroto identify their stemness features,specificity, and growth characteristics. This study was approved by the Institutional Ethics Committee of the First Affiliated Hospital of Dalian Medical University (approval No. LCKY2016-59) on November 18, 2016. Informed consent was obtained from the parturient and family members. hNSCs were seeded at a density of 5 × 105/mL in 25-cm2flasks containing Dulbecco’s modified Eagle’s medium/nutrient mixture F12 (Gibco BRL, Grand Island, NY,USA) supplemented with mitogenic factors, L-glutamine (2 mM; Amresco,Solon, OH, USA), 1% penicillin/streptomycin (Gibco BRL), 2% B27 supplement,1% N2 supplement, 20 ng/mL epidermal growth factor (Gibco BRL), and 20 ng/mL basic fibroblast growth factor (Gibco BRL). The cells were cultured at 37°C under constant humidity in 5% CO2to generate neurospheres (Wang et al., 2017). The growth medium of the NSCs was refreshed every 48 hours.The spontaneous differentiation of NSCs was induced by eliminating basic fibroblast growth factor and epidermal growth factor from the complete medium, and then adding 10% fetal bovine serum.
Human BMECs (hBMECs; RRID: CVCL_4U95) were purchased from ScienCellTMResearch Laboratories (Cat# cAP-0002, supplied by Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd., Shanghai, China), and cultured in Dulbecco’s modified Eagle’s medium/Nutrient mixture F12 supplemented with 1%penicillin/streptomycin, 10% fetal bovine serum, and 2 mM L-glutamine. The cells were incubated in 5% CO2at 37°C and 95% humidity until confluence.
Flow cytometry identification
Flow cytometry was conducted for immunophenotyping of antigens on the NSCs (2ndto 4thpassage) and BMECs (3rdto 5thpassage). The cells were collected, adjusted to 1 × 106/mL, incubated with mouse anti-human nestinfluorescein isothiocyanate (FITC) monoclonal antibody (1:100, BioLegend, San Diego, CA, USA, Cat# 656805, RRID: AB_2566381) (for NSCs identification)and mouse anti-human CD31-FITC monoclonal antibody (1:100, BioLegend,Cat# 303103, RRID: AB_314329) (for BMECs identification) in the dark for 30 minutes, stained, washed twice with phosphate buffer saline (PBS) by centrifugation for 10 minutes at 300 ×g, and analyzed by a FACS Aria II flow cytometer (Becton Dickinson, San Jose, CA, USA). Ten thousand events were counted and analyzed by the FACS DiVa software program (Becton Dickinson).All antibodies and isotype controls were provided by Pharmigen (Becton Dickinson).
Design and preparation of microfluidic device
The microfluidic device for NVU constructionin vitrowas designed as an integrated assembly. The device comprised an upper polydimethylsiloxane(PDMS; Sylgard 184, Dow Corning, Auburn, AL, USA, monomer/crosslinker:10/1 wt%) layer, a microporous polycarbonate (PC) membrane (Costar,Corning, NY, USA), a lower PDMS layer, and a glass slide coated with PDMS.The brain microvascular blood flow was mimicked by the upper PDMS layer,endothelial cells were loaded on the PC membrane to mimic BBB, and a neural unit was constructed by the lower PDMS layer. In the upper and lower PDMS layers, there were both perfusion channels and culture chambers.These two culture chambers were vertically aligned and separated by the PC membrane. Because the upper and lower PDMS layers were identically structured, we fabricated a positive mold using SU-8 negative photoresist(3035, MicroChem, Newton, CA, USA) with an improved soft lithography method (Liu et al., 2014). Microstructural PDMS blocks were then cast with this mold. The PDMS chip contained perfusion channels with a 200-μm height and 100-μm width, and culture chambers with a 2-mm height and 8-mm width. Through-hole inlets, outlets, and culture chambers were generated by punching.
Microfluidic cell seeding and co-culture
The device with cell seeding in the chip was assembled in three steps. First, 48 hours before chip assembly, hBMECs (1 × 106/mL) were seeded and cultured on the sterile PC membrane, then precoated with bovine plasma fibronectin to mimic the vascular endothelial barrier.
Next, the lower PDMS block and PDMS-coated glass slide were subjected to plasma treatment. The surface containing the channel structure in the lower layer and the PDMS surface of the glass slide were irreversibly bonded.Subsequently, only the outlet, inlet and chamber sites were opened for fluid administration. The upper PDMS layer and lower PDMS layer-glass slide were sterilized by ultraviolet light. After plasma treatment, the lower PDMS layerglass slide was sealed with the endothelial cell-loaded PC membrane and upper PDMS chip, respectively. In particular, the cell laden surface of the PC membrane was turned upwards and sealed with the surface containing channel structure in the upper layer chip. The resulting microfluidic device comprised an upper PDMS layer, an endothelial cell-loaded PC membrane,a lower PDMS layer, and a PDMS-coated glass slide from top to bottom. The hydrophilicity of the entire chip was enhanced by plasma treatment, which facilitated fluid flow.
Once the microfluidic device was assembled, continuous perfusion was conducted. Culture medium was first added to the liquid inlets using a pipette to fill the channels and culture chambers, and to remove any air bubbles.Next, using a dual-channel precision syringe pump (LSP02-1B, Baoding Longer Precision Pump Co., Ltd., Baoding, China) and polytetrafluoroethylene tubing (inner diameter 0.76 mm, outer diameter 1.59 mm, IDEX Health &Science, Oak Harbor, WA, USA), culture medium was pumped into the two medium inlets for the perfusion culture at a stable flow rate. Finally, the culture medium of the endothelial cells was introduced to perfuse the upper chamber. A single-cell suspension of NSC neurospheres at an optimized density of 5 × 105/mL was loaded into the lower chip via individual inlets,and adhered to the bottom surface of the culture chamber overnight.Differentiation medium was continuously perfused at 1 μL/min for 7 days in a 37°C incubator with 5% CO2and humidified air, and the perfusion fluids were recycled from the outlets.
Quantitative reverse transcription polymerase chain reaction
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was used to detect markers in samples from the disassembled microfluidic device after being induced by different perfusion times (5, 7, 9, 11 days). Briefly,total RNA was extracted from differentiated and control groups by TRIzol(Takara, Beijing, China), and reverse transcribed into complementary DNA with a PrimeScript™ reverse transcription kit (Takara, Tokyo, Japan). qRTPCR was then performed using a Roche LightCycler480 Real-time System(Roche, Basel, Switzerland) after 2 μL of complementary DNA was mixed with appropriate primers and SYBR® Premix Ex Taq™ II reagent (Takara).The sequences of the primers are: nestin (NSC marker) forward: 5′-GAG AGG GAG GAC AAA GTC CC-3′, reverse: 5′-TCC CTC AGA GAC TAG CGC AT-3′;microtubule-associated protein 2 (MAP2; neuron marker) forward: 5′-GCT ATC CCA GGA CCC CTC AC-3′, reverse: 5′-TCA GCC CCA TGG TCC ACA CG-3′;glial fibrillary acidic protein (GFAP; astrocyte marker) forward: 5′-CTG TTG CCA GAG ATG GAG GTT-3′, reverse: 5′-TCA TCG CTC AGG TCC TT-3′; myelin basic protein (MBP; oligodendrocyte marker) forward: 5′-TTA GCT GAA TTC GCG TGT GG-3′, reverse: 5′-GAG GAA GTG AAT GAG CCG GTT A-3′. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control for normalization. The sequence was designed as GAPDH forward: 5′-CGA CAG TCA GCC GCA TCT T-3′, reverse: 5′-CCA ATA CGA CCA AAT CCG TTG-3′.
Immunofluorescent staining
Immunostaining was performed to characterize the maintenance,proliferation, and differentiation of hNSCs, as well as the maintenance and proliferation of BMECs. The cell samples were treated by 0.2% Triton-X100(MilliporeSigma, St. Louis, MO, USA), 4% paraformaldehyde (MilliporeSigma),and 5% bovine serum albumin in PBS, incubated with primary rabbit antinestin (1:200; Abcam, Cambridge, UK, Cat# ab93666, RRID: AB_1952228),rabbit anti-SOX2 (1:200; Abcam, Cat# ab75627, RRID: AB_1310697), rabbit anti-MAP2 (1:200; Abcam, Cat# ab32454, RRID: AB_1280990), mouse anti-GFAP (1:200; Abcam, Cat# ab86144, RRID: AB_1925029), mouse anti-MBP(1:200; Abcam, Cat# ab62631, RRID: AB_956157), sheep anti-Von Willebrand factor (vWF) (BMEC marker; 1:200; Abcam, Cat# ab8822, RRID: AB_946122),and rabbit anti-zonula occludens-1 (ZO-1; intercellular tight junction marker;1:200; Abcam, Cat# ab59720, RRID: AB_2271824) antibodies overnight at 4°C following the manufacturer’s instructions, rinsed by PBS, and thereafter incubated with goat anti-rabbit IgG-FITC antibody (1:100; MilliporeSigma,Cat# F0382, RRID:AB_631744), and goat anti-mouse IgG-tetramethyl rhodamine antibody (1:100; MilliporeSigma, Cat# T5393, RRID:AB_648996) at room temperature for 90 minutes. Cell nuclei were stained and their positions were identified with 4′,6-diamidino-2-phenylindole staining solution (1:500,MilliporeSigma, Cat# D9542). The imaging settings and quantitative data were identical to those for cell viability evaluation. Fluorescent images were taken by fluorescence microscopy (Leica DMI 4000B, Solms, Hesse-Darmstadt,Germany) equipped with a digital camera (Leica DFC 500).
Assessment of cell viability
To characterize the maintenance and death of BMECs and neuro-glial cells in the microfluidic device, the PDMS layers were detached from each other for sample collection after 7 days of perfusion. The recycled samples were then incubated with calcein (1:1000; LIVE/DEAD® viability/cytotoxicity kit,Molecular Probes, Eugene, OR, USA) and propidium iodide (1:1000) for 20 minutes at room temperature in the dark. Before and after incubation in the staining solution, the samples were washed by PBS twice for 5 minutes eachtime. The neural chamber was observed with a Leica DMI 4000B fluorescence microscope equipped with a Leica DFC 500 digital camera. BMEC samples cultured on the PC membrane were observed under a Leica SP8 laser scanning confocal microscope after being sectioned into 20 layers. Each image was acquired at identical light source intensity and pixel. Quantification was performed with Image-Pro Plus software of the Media Cybernetics IPP 5.0 system (Rockville, MD, USA) by obtaining five individual stacks before projection along the z-axis. To measure the ratio of live cells to dead cells,the fluorescence images were recorded at 480 nm and 590 nm, respectively.The viability was assessed by calculating the percentage of live cells to total cells. The cells in five randomly selected visual fields (100×) were counted using Image Pro-Plus Software (Image Pro-Plus 6.0; Media Cybernetics, Silver Spring, MD, USA).
Evaluation of the neurovascular interface integrity and permeability
To evaluate the integrity of the endothelial barriers between two chambers,a confluent layer of cells in the NVU device was perfused by 25 μM FITCconjugated dextran (4000 Da, Sigma) at 1 μL/min on the upper side. A device that did not contain an hBMECs layer was used as a negative control.Subsequently, the upper chambers were perfused with serum-free medium containing FITC-dextran instead for 6 hours. The perfusion liquid was collected from the lower outlet hourly to assess the permeation amount of FITC-dextran. A calibration curve of the concentration of FITC-dextran versus absorbance at 490 nm was plotted by an EL808 fluorescence microplate reader (BioTek, Highland Park, VT, USA). The intensity was subtracted from the background (serum-free medium). The concentration of the dye permeating the endothelial barrier was obtained through normalization to the calibration curve.
Stimulation of the inflammation via tumor necrosis factor-α
To evaluate the response of the NVU model to inflammatory stimulus, the neurovascular communication functions were biochemically modulated by 2 hours of exposure of the upper chambers (hBMECs) to serum-free medium containing 10 ng/mL tumor necrosis factor-α (TNF-α; Sigma) at 1 μL/min. The upper chambers were then perfused with serum-free medium containing FITC-dextran for 5 hours. The perfusion liquid was collected hourly from the lower outlet to assess the permeation amount of FITC-dextran.
Stimulation of oxygen and glucose deprivation/reperfusion
The upper and lower chambers were rinsed with sterile PBS, and the perfusion fluid was replaced with glucose-free and serum-free Dulbecco’s modified Eagle’s medium. The model was placed in a three-gas incubator at 37°C for 3 hours of anoxic culture, with an anaerobic mixture of 1% O2, 5%CO2, and 94% N2(volume fraction). After the oxygen and glucose deprivation(OGD) treatment, the perfusion fluid of both chambers was replaced with conventional complete medium, and the model maintained oxygen and glucose reperfusion in an incubator with saturated humidity and 5% CO2at 37°C. For assessing the level of cell death after OGD/R, the release of intracellular lactate dehydrogenase (LDH) into the perfusate was measured with a LDH Assay Kit (Solarbio, Beijing, China) according to the manufacturer’s instruction (Ma et al., 2019), where LDH (U/mL) =y/T×103(ywas the concentration of standards, μmol/mL;Twas the reaction time, 15 minutes).The LDH leaking rate and transmission rate of the FITC-labeled dextran in the NVU model were detected at 0, 6, 12, 24, and 48 hours. The control group was also detected at the same time points.
Statistical analysis
Quantitative data were expressed as mean ± standard deviation (SD).Statistical analysis was performed according to Student’st-test using SPSS 18.0 (SPSS, Chicago, IL, USA). Each experiment was performed at least in triplicate.P< 0.05 was considered statistically significant.
Results
Characterizations of neural stem cells and brain microvascular endothelial cells
Primary NSCs and BMECs were characterized before seeding into the microfluidic device. In thein vitroculture of hBMECs in the fusiform or polygon, cells were closely arranged without overlap, with a single pebble sample structure (Figure 1A), and they stably expressed endothelial cellspecific proteins vWF (Figure 1B). Flow cytometry results (Figure 1C)showed that hBMECs stably expressed the platelet endothelial cell adhesion molecule-1 (CD31, positive rate 98.9 ± 1.5%). The 2ndto 5thgenerations of hBMECs were selected for follow-up research.
After primary culture for 2-3 days, the single cells aggregated into spheres,which were small and irregular in shape. On the 5thday of culture, the suspended spheres of hNSCs had uniform shape and good refraction, and after cultivating the neurospheres for 7 days, the cells were viable with a bright, round shape (Figure 2A) and they stably expressed SOX2 (Figure 2B). The flow cytometry results (Figure 2C) showed that the hNSCs stably expressed the specific nestin protein (positive rate 96.7 ± 2.2%), which conformed the characteristics of the NSCs. The 2ndto 5thgenerations of hNSCs were selected for follow-up research.
Composition of the microfluidic neurovascular unit model

Figure 1 |Identification of human brain microvascular endothelial cells.(A) The in vitro morphology of the brain microvascular endothelial cells was a monolayer with typical paving stone-like shapes. (B) The brain microvascular endothelial cells cultured in vitro for 7 days were stained with von Willebrand factor (green, fluorescein isothiocyanate) and the nuclei were stained with 4′,6-diamidino-2-phenylindole (blue).Scale bars: 200 μm (left) and 100 μm (right). (C) The expression of CD31 in the brain microvascular endothelial cells was analyzed by flow cytometry.
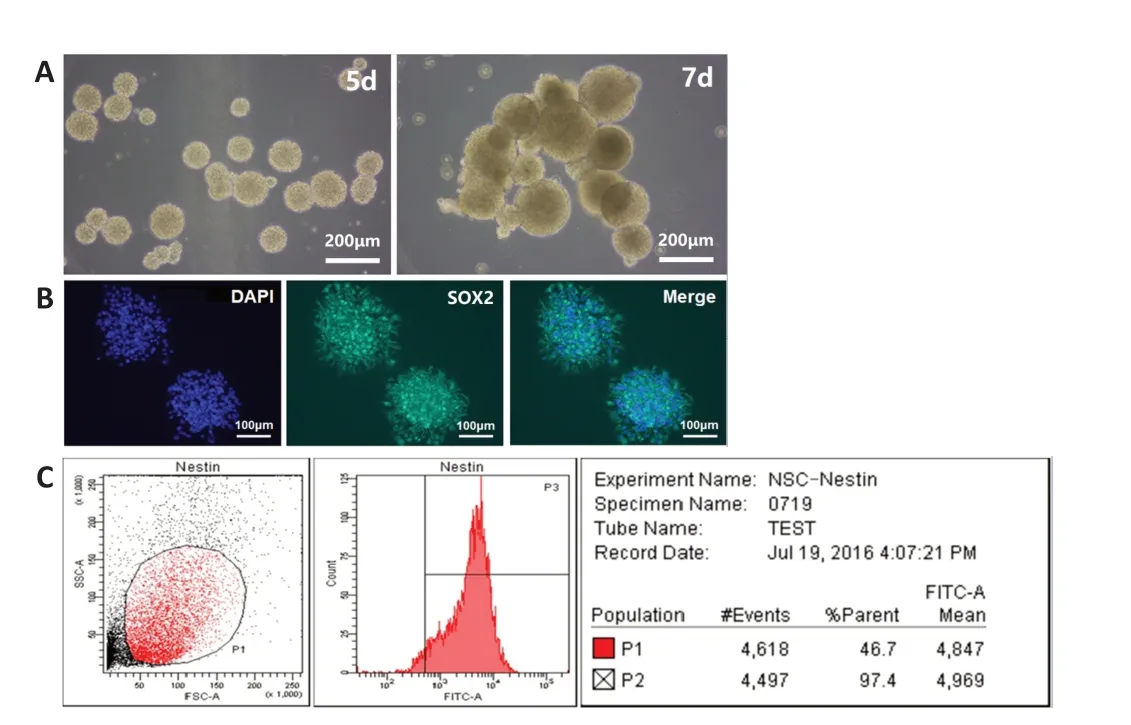
Figure 2 |Identification of human neural stem cells.(A) The primary human neural stem cells formed dense spheres with regular shape, good refraction, and smooth edges in vitro. (B) The primary neural stem cells cultured for 7 days were stained with SOX2 (green, fluorescein isothiocyanate) and the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; blue). Scale bars: 200 μm in A; 100 μm in B. (C) The expression of nestin in the neural stem cells was analyzed by flow cytometry.
The NVU model was divided into various functional areas of the integrated composite structure. As shown in Figure 3A, the microfluidic chip comprises upper PDMS layer 1, PC membrane 2, lower PDMS layer 3, and bottom glass substrate 4. The upper PDMS layer 1 included liquid inlet 11 penetrating the upper PDMS layer 1 and communicating with 31, liquid outlet 12 penetrating the upper PDMS layer 1 and communicating with 32, blood vessel unit liquid inlet 13, blood vessel unit liquid outlet 14, and an upper PDMS layer culture chamber 15 for BMEC culture. Lower PDMS layer 3 included neuro-glial unit inlet 31, neuro-glial unit outlet 32, and lower PDMS layer culture chamber 33 for NSC inoculation. Upper chamber 15 and lower chamber 33 were aligned in the vertical direction, which seamlessly clamped to polycarbonate film 2. The culture medium filled the inlets (13, 11-31), microfluid channels, cell culture rooms (15, 33), and waste liquid ports (14, 12-32). The inlets were connected to the injection pump through a hose, and the microfluidic channels were closed inside the microfluidic chip. The cell culture rooms, liquid inlets, and waste liquid ports were connected through microfluidic channels. Figure 3B showed the top view of the chip. Figure 3C was the combined diagram of the chip model. The real object is shown in Figure 3D.
hNSCs can be induced into a variety of cell types, such as neurons, astrocytes,and oligodendrocytes, which are typically difficult to culture and proliferatein vitro(Figure 4A). As shown in Figure 4B, the entire microfluidic device comprised a neural chamber (for NSCs culture) and microfabricated vascular chamber (for endothelial cells culture). The two chambers were separated by a microporous PC membrane with a 10-μm thickness and 0.4-μm pore size, which was used to mimic BBB by loading hBMECs. The two chambers were fabricated with PDMS (weight ratio to curing agent, 10:1), which has been widely applied to microfluidic platform fabrication for biological studies because of its high gas permeability and biocompatibility. There were independent inlets and outlets in each chamber (1-mm width). The microchannels were connected with a syringe pump to drive culture medium flow. The flow rate was adjusted to 1 μL/min, which approaches that of microvesselsin vivo.
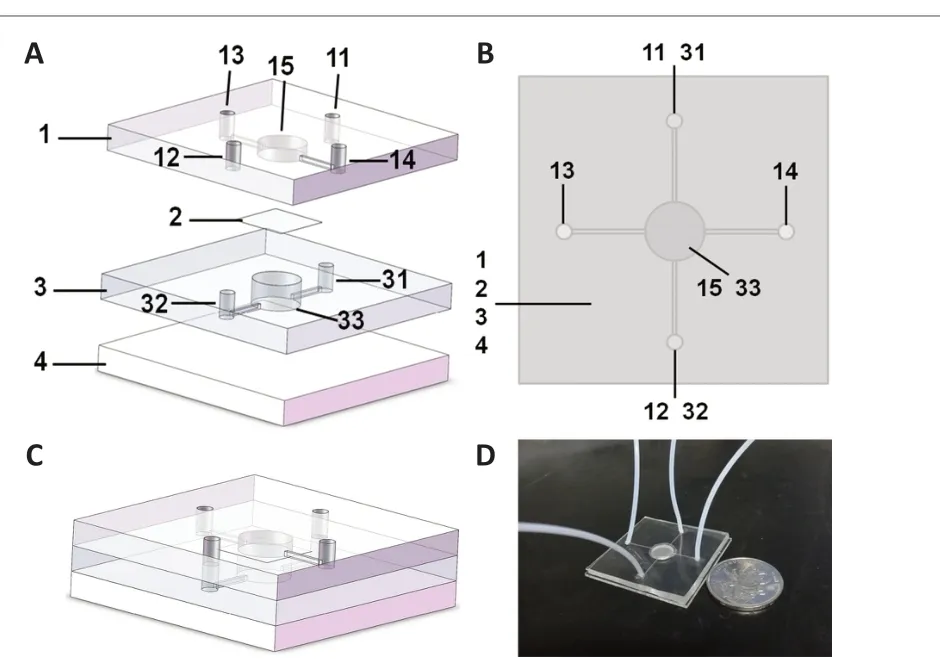
Figure 3 |Model design of the neurovascular unit-on-a-chip.(A) Model decomposition of the neurovascular unit-on-a-chip. The microfluidic chip comprises upper polydimethylsiloxane (PDMS) layer 1, microporous polycarbonate membrane 2, lower PDMS layer 3, and bottom glass substrate 4. The upper PDMS layer 1 included liquid inlet 11 penetrating the upper PDMS layer 1 and communicating with 31, liquid outlet 12 penetrating the upper PDMS layer 1 and communicating with 32,blood vessel unit liquid inlet 13, blood vessel unit liquid outlet 14, and upper PDMS layer culture chamber 15 for brain microvascular endothelial cell culture. The lower PDMS layer 3 included neuro-glial unit inlet 31, neuro-glial unit outlet 32, and lower PDMS layer culture chamber 33 for neural stem cells inoculation. (B) Top view of the neurovascular unit-on-a-chip. (C) Model combination diagram of the neurovascular unit-on-a-chip. (D)A photograph of the neurovascular unit-on-a-chip model.
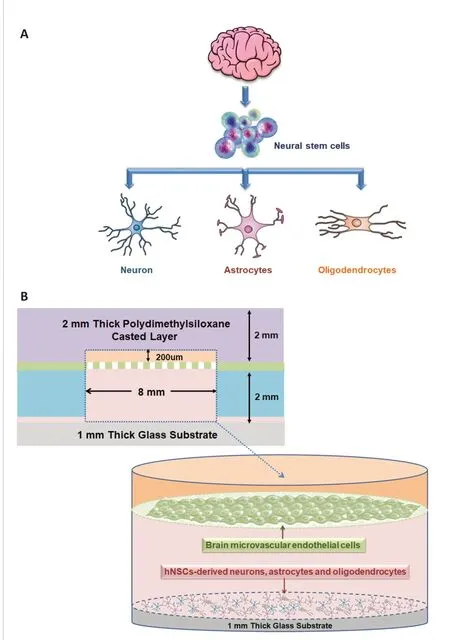
Figure 4 |Cell culture in the neurovascular unit model.(A) The construction principle of the neural unit in the neurovascular unit model. (B) Side view of the cell culture chambers and cell inoculation of the neurovascular unit model.
Neural and glial sub-populations from human NSCs
qRT-PCR was used to detect the gene expression of hNSCs differentiated from the NVU model after being induced for different perfusion times. As shown in Figure 5A, the expression level of nestin in NSC-derived cells gradually decreased with the induction time until it reached the lowest level on the 11thday. The expression of neuron gene MAP2 (Figure 5B) and astrocyte gene GFAP (Figure 5C) reached a maximum on the 7thday of induction, and then decreased gradually. The expression level of the oligodendrocyte gene MBP (Figure 5D) gradually increased with the induction time, and reached the highest level on the 11thday. Because the neurons and astrocytes are important components of the human brain, 7 days was chosen as the optimal induction time of NSCs in the NVU models.
After staining with selective markers MAP2, GFAP, and MBP (Additional Figure 1), GFAP was significantly expressed in the NSC-derived cells under dynamic conditions, confirming the ubiquity of astrocytes (Figure 5F and H). NSCderived neurons existed in patches, varying significantly in different samples(Figure 5E and H). There were small quantities of oligodendrocytes in these cultures (Figure 5G and H).
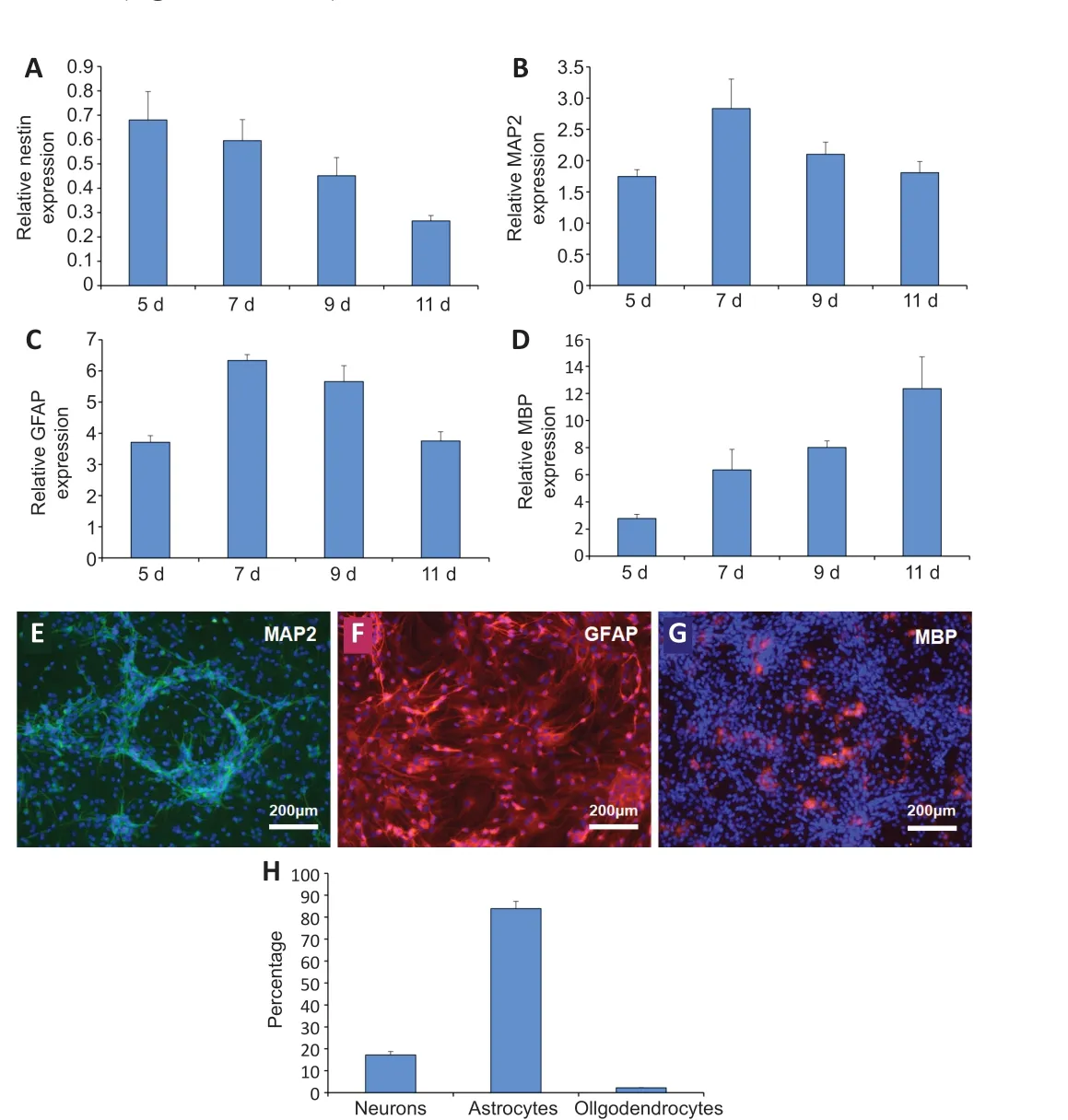
Figure 5 |Comparison of the neuroglial differentiation of neural stem cells with different induction times using a quantitative real-time polymerase chain reaction.(A-D) Gene expression was detected by quantitative real time polymerase chain reaction. (A) The expression of nestin in the differentiated cells derived from neural stem cells at different induction times. (B) The expression of microtubule-associated protein 2 (MAP2) in the differentiated cells derived from neural stem cells at different inductiontimes. (C) The expression of glial fibrillary acidic protein (GFAP) in the differentiated cells derived from neural stem cells at different induction times. (D) The expression of myelin basic protein (MBP) in the differentiated cells derived from neural stem cells at different induction times. The target gene expression was normalized to glyceraldehyde-3-phosphate dehydrogenase. (E) The differentiation of neural stem cells into neurons in the neurovascular unit model after 7 days of induction. MAP2-positive cells showed green fluorescence after fluorescein isothiocyanate staining and nuclei staining showed blue fluorescence under 4′,6-diamidino-2-phenylindole. (F) The differentiation of neural stem cells into astrocytes in the neurovascular unit model after 7 days of induction. GFAPpositive cells showed red fluorescence after tetraethyl rhodamine isothiocyanate staining and nuclei staining showed blue fluorescence under 4′,6-diamidino-2-phenylindole.(G) The differentiation of neural stem cells into oligodendrocytes in the neurovascular unit model after 7 days of induction. MBP-positive cells showed red fluorescence after tetraethyl rhodamine isothiocyanate staining and nuclei staining showed blue fluorescence under 4′,6-diamidino-2-phenylindole. Scale bars: 200 μm. (H) Neuro-glial sub-population percentages of neural stem derived cells showing the relative distribution of neurons to glia after 7 days of induction culture. Data are expressed as mean ± SD and were analyzed by a Student’s t-test. The experiments were repeated three times.
Morphology and viability of the cells on the microfluidic device
After 7 days of perfusion culture, hBMECs were attached to the microporous PC membrane, forming a monolayer barrier mimicking BBBin vivo. The hBMECs were cobblestone-shaped and tightly in contact, producing a complete endothelium (Figure 6A). A single-cell suspension of NSCs was loaded into the lower chambers through independent inlets, and differentiated into neuro-glial cells stably after 7 days of induction.NSCs-derived neurons and glial cells were completely confluent and indistinguishable (Figure 6B). After culture in respective chambers, the viabilities of each cell component were determined by calcein-potassium iodide double staining (Figure 6C and D). More than 90% of the endothelial and neuro-glial cells were live, significantly exceeding the number of dead cells (P< 0.05; Figure 6E and F).
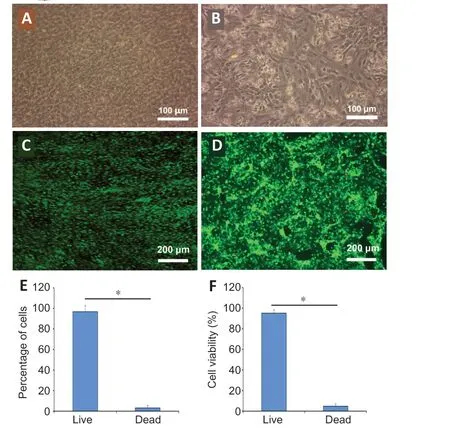
Figure 6 |Growth of the brain microvascular endothelial cells and neural stem cell derived neuro-glial cells in the model of the neurovascular unit.(A) The brain microvascular endothelial cells were uniform in size and grew on the polycarbonate membrane in the NVU model. (B) The neural stem cell derived neuroglial cells appeared as a monolayer with spindle morphology in the NVU model. (C) The survival of the brain microvascular endothelial cells on the polycarbonate membrane in the NVU model. (D) The survival of neural stem cells derived neuro-glial cells in the NVU model. The living cells showed green fluorescence after calcein staining and the dead nuclei showed red fluorescence under propidium iodide. Scale bars: 100 μm in A and B;200 μm in C and D. (E) The survival rate of the brain microvascular endothelial cells in the NVU model. (F) The survival rate of neural stem cell derived neuro-glial cells in the NVU model. Data are expressed as mean ± SD. *P < 0.05 (Student’s t-test). The experiments were repeated three times.
Integrity and permeability of the neurovascular unit
After 7 days of culture through perfusion, the integrity of the endothelial barrier in the vascular unit was detected by immunofluorescence staining.hBMECs in the chambers expressed vWF in the normal culture (Figure 7A),verifying that the microvascular endothelial activity was normal. Under the laser confocal microscope, the positive expression of ZO-1 protein was detected among hBMECs (Figure 7B), indicating that tight junctions were formed between endothelial cells and the integrity of the barrier was established.To determine permeability of the endothelial barrier, 4 kDa FITC-dextran was introduced to the prepared device. The upper channels were completely perfused with medium containing FITC-dextran. The perfusion liquid from the basolateral chamber outlet was detected hourly. According to the FITCdextran calibration curve (only PC membrane, without cells), the absorbance increased rapidly, and the equilibrium state was reached in 1 hour. Moreover,the fluorescence intensity of the fully confluent endothelial barrier increased with time, reaching a steady state in 4 hours and remaining stable thereafter.As shown in Figure 7C, the permeation rate of dextran in the NVU model was significantly less than that in the control group (P< 0.01). hBMECs remarkably decreased the barrier permeability, as proven by the lower FITC-dextran intensity of the basolateral chamber.
Biochemical modulation of the neurovascular unit
The influence of TNF-α, an inflammatory agent, on the microenvironment of the NVU model was evaluated. The vascular layer in which the endothelial barrier was not exposed to TNF-α was used as a negative control. After stimulation of TNF-α for 2 hours, FITC-dextran leakage crossing the neurovascular interface remarkably increased in the sample with TNF-α circulation, resulting in an increase in fluorescence intensity of the perfusion liquid from the basolateral chamber. Therefore, TNF-α, which was circulated from the vascular side, attenuated the endothelial barrier integrity that was greater in the TNF-α-free sample (Figure 8A), and likely activated cells in both chambers, inferring that the co-cultures in the independent compartments communicated. It is postulated that the tight endothelial barrier was disrupted because the endothelium became inflamed via the well documented proinflammatory action of TNF-α duringin vivoneuroinflammation.
Finally, to mimic the process of cerebral ischemia reperfusion injuryin vitro, we screened OGD/reperfusion (OGD/R) injury conditions based on the functional NVU model. After OGD for 3 hours, the alteration of the neurovascular interface integrity and LDH activity of perfusion liquid from the lower outlet were detected at 0, 6, 12, 24, and 48 hours of reperfusion.After reperfusion, the permeability of the endothelial barriers in NVU model increased after 12 hours of reperfusion, and reached a maximum at 24 hours and 48 hours of reperfusion (Figure 8B). The activity of LDH began to increase at 6 hours of reperfusion and reached a maximum at 24 hours(Figure 8C). The degree of damage of OGD/Rin vitrocan be quickly realized by the changes of LDH activity in the perfusion fluid, which is sensitive and convenient. The injury of OGD/R on the activity of the microvascular endothelial cells and neuro-glial cells in the NVU model is shown in Additional Figure 2. The survival rates of neuro-glial cells and hBMECs began to decline at 6 and 12 hours of reperfusion, respectively, and reached the lowest survival rate at 24 and 48 hours of reperfusion.

Figure 7 |Neurovascular interface integrity quantification and tight junction expression across confluent endothelial barriers in the neurovascular unit model.(A) The expression of von Willebrand factor (vWF) in the endothelial barrier. vWF-positive cells showed green fluorescence after fluorescein isothiocyanate (FITC) staining and nuclei staining showed blue fluorescence under 4′,6-diamidino-2-phenylindole. (B) The expression of zonula occludens-1 (ZO-1) in the endothelial barrier. The tight junctions between cells showed green fluorescence after FITC staining and nuclei staining showed blue fluorescence under 4′,6-diamidino-2-phenylindole. Scale bars: 100 μm in A; 25 μm in B. (C) The permeability of neurovascular unit models with a human brain microvascular endothelial cell (hBMEC) barrier and a control group with FITC-labeled dextran. The fluorescence intensity was measured by optical density (OD). Data are expressed as mean ± SD, and were analyzed by Student’s t-test. The experiments were repeated threetimes.

Figure 8 |Effects of tumor necrosis factor-α (TNF-α) and oxygen and glucose deprivation/reperfusion on the integrity and permeability of the neurovascular unit model.(A) Alterations of the neurovascular interface integrity between groups with and without TNF-α stimulation. (B) Permeability changes of endothelial barriers between the control group and groups with different reperfusion time points after 3 hours of oxygen and glucose deprivation. The fluorescence intensity was measured by optical density (OD) in A and B. (C) The alterations of lactate dehydrogenase (LDH) activity between the control group and groups with different reperfusion time points after 3 hours of oxygen and glucose deprivation. Data are expressed as mean ± SD. *P < 0.05. #P < 0.05, ##P < 0.01,vs. control group; †P < 0.05, ††P < 0.01, vs. 0 h group (Student’s t-test). The experiments were repeated three times.
Discussion
Using microfabricated devices to mimic physiological structures, functions,and microenvironments requires the culture of diverse cells in devices with complex architectures and several compartments that are vertically or horizontally stacked (Phan et al., 2017; Ahadian et al., 2018). However, it is still challenging to culture more than two types of cells in these microdevices(Young and Beebe, 2010; Zervantonakis et al., 2011; Yan et al., 2021). First,it is rather difficult to seed cells in sealed chambers because of placement and viability. Second, continuous replenishment of fresh medium during cell growth in sealed systems is significantly hampered. Finally, if tissue vascularization is needed, the differentiation or growth rates of vascular and parenchymal cells should be optimized to expand the assay window. Particular attention should be paid to the process if there are more than three types of cells (e.g., our neuro-glial co-cultures) along the vascular endothelial barrier in a culture system.
Cell lines have been widely used to establishin vitromodels because of their immortalization and easy culture characteristics (Cucullo et al., 2007; Ravi et al., 2015). However, the data obtained from cell lines to buildin vitromodels are quite different from the real situation of the human body. Because the limitations of nonhuman or immortalized cell lines are well recognized,researchers have increasingly isolated and cultured primary human cells for experimental research (Ghosh et al., 2011). Since Reynolds and Weiss (1992)isolated NSCs from the adult mammalian forebrain, new neurons have been confirmed to form in adulthood. As a result, lifelong neurogenesis has been demonstrated in most mammals (e.g., human) (McKay, 1997; Eriksson et al., 1998). NSCs are now generally considered multipotent and capable of unlimited self-renewal, differentiation into neurons, oligodendrocytes and astrocytes, as well as having persistent existence in the CNS (Grochowski et al., 2018; Ludwig et al., 2018). Under both pathological and physiological conditions, the balance between NSC differentiation and maintenance plays a key role in providing specific neural populations for the brain. Moreover,harvesting and proliferating neurons and glial cellsin vitroremain difficult.NSCs are easier to culture and expandin vitrothan any other kind of cells in the CNS. Our previous work demonstrated that NSCs still maintain selfrenewal and differentiation after 20 generations of expansion, and they are a good candidate for tissue engineering research (Wang et al., 2017).
In the NVU model device, co-culture was stable and easy, and the cultured cells in both chambers survived for a long time. In the modular neural chamber, NSCs were differentiated into neurons, oligodendrocytes, and astrocytes in the presence of serum using a previously established method(Wang et al., 2017). The relative populations of these NSCs-derived neuroglial cells were closely related to the time of induction and the amount of serum in the culture medium (Lippmann et al., 2011). To maximize the differentiation efficiency and minimize the variability of the cellular components, NSCs were cultured by adding 10% fetal bovine serum without epidermal growth factor or basic fibroblast growth factor. Additionally, the capacity of NSCs to differentiate into three types of neuroglial cells reached a maximum 7 days after induction. In this device, NSCs were induced to stably differentiate into astrocyte-based neuroglial units. The percentages of the types of neuroglia did not completely resemble thein vivocells (Cohen-Kashi Malina et al.,2009); as a result, further studies on the reconstruction of neuroglial cells by applying inducing factors are ongoing in our group.
The neurovascular interface in our device was built using primary hBMECs in which the tight junction protein ZO-1 was expressed. The permeation of FITCdextran was also successfully monitored by our NVU device in real time, which was impossible using transwell chambers. As evidenced by the lower FITCdextran intensity of the basolateral chamber, hBMECs remarkably decreased the barrier permeability, indicating that tight junctions formed in the NVU model device. In addition, the endothelial barrier after TNF-α stimulation leaked more FITC-dextran than the unstimulated barrier. Accordingly, TNF-α may augment barrier leakage by triggering a pro-inflammatory reaction.Furthermore, we used the NVU model to reproduce OGD/R to mimic hypoxic/ischemic encephalopathy, which accounts for approximately 85% of all cerebrovascular diseases. The results suggested that our NVU model showed good biocompatibility and neurovascular interface integrity, as well as good response to physical and chemical stimulus. However, the modular device was not integrated with a trans-endothelial electrical resistance (TEER) electrode as shown by Griep et al. (2013), aiming to simplify the fabrication process.
The NVU model device in this study had the following advantages. (1) By combining an assembled, integrated, 3D dynamic perfusion microfluidic device with primary human-derived cells (rather than genetically engineered cell lines or animal cells), we developed a reliable, versatile, easily operatedin vitromodel to establish brain structural units that mimicked thein vivocellular microenvironment. (2) Using the differentiation potential of NSCs to obtain three kinds of neuro-glia cells (neurons, astrocytes, and oligodendrocytes),which cannot be readily extracted and proliferatedin vitro. This system is beneficial to the construction of complex NVU using simple methods and minimal cell types. (3) The scarcity of human source cells from primary extraction can be solved by simple chip modules and less cell consumption.The cell survival rate can be increased by continuous perfusion and dynamic culture. (4) This system inspires further construction of pathological models of various neurological diseases, such as hypoxic-ischemic brain damage, and Alzheimer’s disease, allowing for drug safety and efficacy evaluation, dose screening, and individualized medication guidance.
The primary purpose of thein vitroNVU microdevice is to model the basic structural and functional units, as well as the physiological microenvironment,of the human brain. The device is also applicable to predictive screening,assessment, and optimization of drug candidates for BBB permeation, as well as clarifying neurovascular dysfunction for different neurological diseases.Combining this platform with other detection strategies is conducive to the development of CNS therapeutics and individualized medication regimens.However, there are some limitations of the current model. First, the NVU system did not introduce pericytes, which are known to augment the neurovascular interface integrity (Birbrair, 2018; Payne et al., 2019). Second,the neuron to astrocyte to oligodendrocyte ratio (17%: 84%: 2%) in our cultures do not reflect thein vivoratio (Allen and Barres, 2009; Trujillo-Estrada et al., 2019). However, this limitation could be overcome by the convenient design of our device that is amenable to adding directional inducers via perfusate. Third, the current system lacked a TEER detecting device to measure the electrical resistance. In the future, we will introduce the insertion ports of electrode wires into the prepared modular chips to monitor TEER dynamically (Booth and Kim, 2012). The integrity of the neurovascular interface can be monitored dynamically by the TEER sensors over time. We also intend to construct 3D neuronal-glial models using biomaterials (e.g., silk fibroin, alginate, and gelatin) as scaffolds on the basis of previous research(Guo et al., 2015; Liu et al., 2015; Lv et al., 2017), continuously improving the spatial structure and function of the NVU model to better mimic the brain microenvironment.
In summary, we successfully established anin vitrobrain NVU model based on the differentiation potential of NSCs and integrated characteristics of the microfluidic chip technology. In addition to facile seeding and maintenance of the various cells together, with low sample requirements, the designed chip enabled building of complex models in a simple way (at least four types of cells were obtained using only two cell types). Furthermore, the endothelial barriers of the NVU model could mimic the role of BBB and have a good response to inflammatory stimulation. Additionally, the compartmentalized chip may be able to deliver hormones, cytokines, drugs, nutrients, viral/bacterial agents, and exosomes via vascular channels, and NVU can be regarded as a whole system. We will use this microfluidic approach to study the pathogenesis of neurodegenerative diseases, CNS-related drug screening,biodefense, and individualized medication.
Acknowledgments:The authors would like to thank Jing-Yun Ma (Medical College, Ningbo University, China) for her great help in microfluidic technology.
Author contributions:Study conception and design, data analysis and manuscript writing: WJW; experiment implementation: WJW, YCW, WGC;manuscript revision: XG, JL. All authors read and approved the final version of the manuscript.
Conflicts of interest:None declared.Editor note: JL is an Editorial Board member of Neural Regeneration Research.She was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal’s standard procedures, with peer review handled independently of this Editorial Board member and their research groups.
Availability of data and materials:All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Kasum Azim, Universitatsklinikum Dusseldorf, Germany.
Additional files:
Additional file 1:Open peer review report 1.
Additional Figure 1:Differentiation of neural stem cells into neuro-glial cells in the model of neurovascular unit after 7 days of perfusion culture.
Additional Figure 2:Effect of oxygen and glucose deprivation/reperfusion on the activity of brain microvascular endothelial cells and neuro-glial cells in neurovascular unit models.

- 中国神经再生研究(英文版)的其它文章
- Functional in vivo assessment of stem cell-secreted prooligodendroglial factors
- iGluR expression in the hippocampal formation, entorhinal cortex,and superior temporal gyrus in Alzheimer’s disease
- Exploiting Caenorhabditis elegans to discover human gut microbiotamediated intervention strategies in protein conformational diseases
- N-methyl-D-aspartate receptor functions altered by neuronal PTP1B activation in Alzheimer’s disease and schizophrenia models
- Aminopeptidase A and dipeptidyl peptidase 4: a pathogenic duo in Alzheimer’s disease?
- Ubiquitin homeostasis disruption,a common cause of proteostasis collapse in amyotrophic lateral sclerosis?