
Cranial irradiation impairs intrinsic excitability and synaptic plasticity of hippocampal CA1 pyramidal neurons with implications for cognitive function
2022-03-09 07:31MinYiWuWenJunZouPeiYuYuhuaYangShaoJianLiQiangLiuJiatianXieSiQiChenWeiJyeLinYameiTang
中国神经再生研究(英文版) 2022年10期
Min-Yi Wu, Wen-Jun Zou, Pei Yu, Yuhua Yang, Shao-Jian Li, Qiang Liu,Jiatian Xie, Si-Qi Chen, Wei-Jye Lin, Yamei Tang,4,6,*
Abstract Radiation therapy is a standard treatment for head and neck tumors. However, patients often exhibit cognitive impairments following radiation therapy.Previous studies have revealed that hippocampal dysfunction, specifically abnormal hippocampal neurogenesis or neuroinflammation, plays a key role in radiation-induced cognitive impairment. However, the long-term effects of radiation with respect to the electrophysiological adaptation of hippocampal neurons remain poorly characterized. We found that mice exhibited cognitive impairment 3 months after undergoing 10 minutes of cranial irradiation at a dose rate of 3 Gy/min. Furthermore, we observed a remarkable reduction in spike firing and excitatory synaptic input, as well as greatly enhanced inhibitory inputs,in hippocampal CA1 pyramidal neurons. Corresponding to the electrophysiological adaptation, we found reduced expression of synaptic plasticity marker VGLUT1 and increased expression of VGAT. Furthermore, in irradiated mice, long-term potentiation in the hippocampus was weakened and GluR1 expression was inhibited. These findings suggest that radiation can impair intrinsic excitability and synaptic plasticity in hippocampal CA1 pyramidal neurons.
Key Words: GABA-mediated hyperfunction; GluR; intrinsic excitability; long-term potentiation; radiation-induced cognitive impairment; spontaneous excitatory postsynaptic currents; spontaneous inhibitory postsynaptic currents; synaptic plasticity; type I vesicular glutamate transporter; vesicular GABA transporter;whole-cell patch clamp recording
Introduction
Radiation therapy is included in established therapeutic protocols used to treat multiple types of head and neck tumors (McTyre et al., 2013; Owonikoko et al., 2014). While cranial radiotherapy has been proven to significantly extend the survival rate of cancer patients, the treatment is routinely associated with serious complications, including cognitive impairment. Indeed,6 months to 1 year after radiation, 50-90% of patients exhibit cognitive dysfunction that severely affects their quality of life (Greene-Schloesser et al., 2013; Makale et al., 2017). However, the mechanisms by which radiation induces cognitive dysfunction have not been thoroughly elucidated.
The hippocampus has long been considered a pivotal brain area for learning and memory (Bartsch and Wulff, 2015; Wang et al., 2020, 2021; Xue et al.,2021). Structural and functional changes in the hippocampus can result in increased vulnerability to pathological states associated with cognitive deficits(Galvin et al., 1999; von Oertzen et al., 2002; Blum et al., 2012). Notably,
patients receiving brain irradiation exhibited deficits in learning and spatial processing, which are related to hippocampal function (Gondi et al., 2010),while hippocampus-avoidance radiotherapy has been found to preserve cognitive function (Andreas and Kundapur, 2015; Brown et al., 2020). Several studies have reported that the hippocampus is vulnerable to radiation,and have linked radiation-induced structural changes in the hippocampus to cognitive decline (Galvin et al., 1999; Rao et al., 2011; Son et al., 2015).Researchers have also reported that deficits in hippocampal neurogenesis(Monje et al., 2002; Zou et al., 2012) and neuroinflammation (Peng et al., 2014;Montay-Gruel et al., 2019) played critical roles in radiation-induced cognitive impairment (Son et al., 2015). However, radiation-induced electrophysiological adaptation in hippocampal neurons has not been well characterized.
Here, we investigated the long-term impact of a single radiation dose of 30 Gy on the intrinsic electrophysiology and synaptic plasticity of hippocampal CA1 pyramidal neurons. Our findings provide new insights regarding the pathogenic mechanisms underlying radiation-induced cognitive deficits.
Materials and Methods
Animals
Adult 8- to 10-week-old C57BL/6J special pathogen free-level male mice (n=40) weighing 20-25 g were used for all experiments. The mice were obtained from Guangdong Medical Laboratory Animal Center (Guangzhou, China;license No. SCXK (Yue) 2021-0029). All mice were maintained on a 12-hour light-dark circadian cycle and had free access to food and water in a suitable environment with a temperature of 18-22°C and humidity of 50-60%. The mice were monitored on a daily basis and weighed every 7 days to ensure that the experimental intervention was well tolerated. This study was approved by the Animal Research Ethics Committee of Sun Yat-sen University (approval No.Bei-B2019-0218QX) on October 21, 2019. All animal care and experimental protocols were conducted following the institutional guidelines of Sun Yatsen University and the NIH Guide for the Care and Use of Laboratory Animals.Endeavors were made to limit the pain and suffering of the animals used in this study.
Cranial irradiation
The protocol for irradiation of mice has been previously described (Xu et al.,2015). Briefly, adult male mice were separated randomly into control and radiation groups. Cranial irradiation of the mice was performed using a 6 MV β-ionizing-ray linear accelerator (Siemens, Munich, Germany). Anesthetized mice were fixed on a custom-designed platform. The mice were positioned such that the treatment field extended from the post-canthus line to the post-aurem line (Xu et al., 2015). The mice in the radiation group received irradiation in a single dose of 30 Gy, delivered at a rate of 3 Gy/min for 10 minutes with a source-to-skin distance of 100 cm. To exclude the possible effects of anesthesia, mice from the control group underwent the same anesthesia procedures as those in the radiation group, but did not receive irradiation. A total of 22 mice received irradiation and 18 mice underwent a sham operation, and the mice came from three separate cohorts. Only 16 mice in the radiation group (6 mice died after irradiation, mortality rate = 27%) and 18 mice in the control group were assessed 3 months after irradiation. One cohort of mice was used for behavioral tests and electrophysiological recording, and the other two cohorts were separately used for electrophysiological recording and tissue collection, respectively. All assessments were conducted 3 months after cerebral irradiation in both the radiation and control groups.
Novel object location test
To examine the long-term memory of mice, we carried out the novel object location test according to previous reports (Leger et al., 2013; Glasgow et al., 2020). After handling the mice daily for 5 days, they were habituated to a square chamber (33 cm × 33 cm × 20 cm) containing no objects for 10 minutes (day 1). On day 2, the animals were exposed to two identical objects in the chamber and given a total exploration time of 10 minutes. On day 3,one of the two objects was relocated to a new position and the animals were allowed to freely explore the chamber for a total of 10 minutes. Mice that did not explore the two objects for at least 20 seconds within the 10-minute period on day 2 or 3 were removed from the analysis. Throughout the entire experiment, an overhead camera was used to record the behavior of the mice in the chamber. The amount of time spent engaged in exploration on days 2 and 3 was measured by a highly experienced observer (JX) who was blinded to the experimental groups. The experimental chamber and objects were cleaned with 70% ethanol to reduce the influence of mouse odor between the trials. The exploration times were defined as the total time during which the mice sniffed each object (2 cm within the object with the nose angled directly toward the object). Recognition ratios were calculated as the amount of time spent sniffing an object, divided by the total time spent exploring both objects.
Morris water maze
To examine the spatial memory of mice, we performed the Morris water maze (MWM) as previously reported (Vorhees and Williams, 2006). A circular pool (110 cm in diameter) was filled with water that was made opaque using non-toxic white paint. The temperature of the pool was set at 20-22°C.In the training phase, which took 5 days, the mice were trained to find a round platform that was submerged 1 cm below the surface of the water in the northeast quadrant of the pool. Each mouse completed four trials per day. The starting point for each trial was either the southeast or northwest quadrant, selected at random. The search time for each trial was 60 seconds.Mice were guided to the platform if they did not find it within 60 seconds,and were allowed to remain on the platform for 15 seconds after each trial.The mice were allowed to rest between the trials for 1 minute. A probe test,in which the platform was absent from the pool, was carried out on day 6. For the probe test, mice were placed in the pool at a novel location (southwest quadrant) and permitted to swim for 60 seconds. We used TopScanTM2.0(CleverSys, Washington, DC, USA), which is a tracking system with a camera,to calculate escape latency, swimming speed, the percentage of time spent in each quadrant, and the number of crossings into each quadrant. In a cued test, which took place after the probe test, a visible flag was attached to the platform. This allowed us to record the swimming latency for a visible platform.
Electrophysiological recording
The procedures for brain slice preparation and electrophysiological recording were as previously reported (Zou et al., 2020). In brief, mice were quickly decapitated under 1% pentobarbital anesthesia (100 mg/kg,Sigma, St. Louis, MO, USA; intraperitoneal injection). Brains were dissected into ice-cold oxygenated modified artificial cerebrospinal fluid (in mM:250 sucrose, 26 NaHCO3, 10 glucose, 10 MgSO4, 2 KCl, 1.3 NaH2PO4, and 0.2 CaCl2, incubated with 95% O2and 5% CO2). Coronal hippocampal slices (300 μm thick) were prepared using a vibratome (Leica, Heerbrugg,Germany, VT-1200S) and incubated at 34 °C for half an hour in oxygenated regular artificial cerebrospinal fluid (in mM: 126 NaCl, 26 NaHCO3, 10 glucose, 3 KCl, 2 CaCl2, 1.25 NaH2PO4, and 1 MgSO4). After incubation,the brain slices were placed in a recovery chamber at 25 ± 1°C for one hour and then transferred to the electrophysiological recording area.All extracellular solutions were constantly oxygenated using 95% O2/5%CO2. During recording, brain slices were submerged and superfused by normal, oxygenated artificial cerebrospinal fluid solution (2 mL/min)at 32-34°C. Electrophysiological data were recorded using a MultiClamp700B amplifier and analyzed with PClamp software (Molecular Devices, San Jose,CA, USA), followed by filtering at 2 kHz and digitization at 10 kHz using Digidata 1440 (Molecular Devices).
For LTP recording, field excitatory postsynaptic potentials (fEPSPs) from the CA1 stratum radiatum were recorded following stimulation of Schaffer collaterals using a two-concentric bipolar stimulating electrode (FHC,Bowdoin, ME, USA). We defined the strength of synaptic transmission as the initial (10-60% rising phase) slope of the fEPSPs. LTP was induced via a 100-Hz stimulus train with 50 pulses, and signals were recorded for 60 minutes.The level of LTP was determined by the average fEPSPs slope during the last 10 minutes of recording after tetanic stimulation.
To measure the intrinsic excitability of CA1 pyramidal neurons, cells were viewed and selected using an upright microscope (ECLIPSE FN1, Nikon,Tokyo, Japan) with a 40× water-immersion objective and infrared differential interference contrast, as well as a digital camera. Borosilicate glass pipettes with a tip resistance of 3-5 MΩ were prepared using a horizontal pipette puller (P-2000, Sutter Instrument, Novato, CA, USA) and filled with solution(in mM: 105 K-gluconate, 30 KCl, 10 HEPES, 10 phosphocreatine, 4 ATP-Mg,0.3 GTP-Na, and 0.3 glycol-bis-(2-aminoethylether)-N,N,N′,N′-tetraacetic acid(EGTA), pH 7.35, 285 mOsm). Whole-cell recordings were performed using the current-clamp technique, and spikes were induced by injecting a series of depolarizing current pulses in the presence of 20 μM CNQX (a competitive α-amino-3-hydroxy-5-methyl-4-isoxazole-propionicacid (AMPA)/kainate receptor antagonist), 100 μM DL-AP5 (a N-methyl-D-aspartic acid (NMDA)glutamate site antagonist), and 20 μM bicuculline. The threshold of an action potential (AP) was calculated as the depolarization at which the neuron fired.The rheobase was determined by performing a series of current injections and recording the current that elicited the first spike.
For recording spontaneous excitatory postsynaptic currents (sEPSCs), the neuronal membrane potential was held at -70 mV (using the voltage-clamp technique) in 20 μM bicuculline to block gamma-aminobutyric acid type A (GABAA) receptors. For recording spontaneous inhibitory postsynaptic currents (sIPSCs), the neuronal membrane potential was held at 0 mV (using the voltage-clamp technique) in 20 μM CNQX and 50 μM DL-AP5 to block AMPA receptors and NMDA receptors. The pipettes were filled with solution (in mM: 135 Cs-Meth, 10 KCl, 1 MgCl2, 0.2 EGTA, 2 QX-314, 4 ATP-Mg, 0.3 GTPNa, and 20 phosphocreatine, pH 7.3), with an mOsm of 290-300. We did not analyze datasets with a maintenance current > -200 pA or a shift in the value resistance of the series > 20%. Datasets were attained using pClAMP10.7(Molecular Devices), and assessed via Clampfit 10.7® software (Molecular Devices) and Mini Analysis software (Synaptosoft Inc., Leonia, NJ, USA).
Immunofluorescence staining
To examine molecular synaptic plasticity in CA1, the brains were collected from mice after anesthetization via 1% pentobarbital (100 mg/kg,intraperitoneal injection) and intracardiac perfusion with 0.9% saline followed by 4% paraformaldehyde. The brains were post-fixed in 4%paraformaldehyde overnight and then transferred to 20% sucrose in phosphate buffered saline (PBS), followed by 30% sucrose in PBS until they sank to the bottom of the container. We prepared 30 μm-thick coronal sections using a microtome (NX50, Thermo Waltham, MA, USA). Brain slices containing the hippocampus were selected and washed with PBS three times for 5 minutes. After that, sections were incubated with 5% normal donkey serum (Beyotime Biotechnology, Shanghai, China) with 0.4% Triton X-100 for 1 hour at room temperature. After blocking, slices were incubated with a rabbit polyclonal antibody against vesicular GABA transporter (VGAT; 1:500,Cat# 131002, RRID: AB_887871, SySy, Goettingen, Germany) and a polyclonal guinea pig antibody against type I vesicular glutamate transporter VGLUT1;1:500, Cat# 135304, RRID: AB_887878, SySy) in 1% normal donkey serum solution at 4°C overnight. The brain slices were next incubated with donkey anti-rabbit Alexa Fluor 488-conjugated IgG (1:500, Cat# 711-545-152, RRID:AB_2313584, Jackson ImmunoResearch Labs, Philadelphia, PA, USA) and donkey anti-guinea pig Alexa Fluor 594-conjugated IgG (1:500, Cat#106-585-003, RRID: AB_2337442, Jackson ImmunoResearch Labs) for 2 hours at room temperature, washed with PBS three times (5 minutes each), and covered with FluoroshieldTMwith 4,6-diamino-2-phenyl indole (F6057, Sigma).Images were acquired using an LSM 800 confocal microscope (Carl Zeiss,Oberkochen, Germany) with a 20× objective.
Western blot assay
The electrophysiological recordings revealed a significant change in synaptic plasticity. Accordingly, we analyzed changes in the expression of synapseassociated markers, including neurotransmitter vesicular transporters (VGAT and VGLUT1) and neurotransmitter receptors including AMPAR (GluR1 and GluR2), NMDAR (NMDAɛ2), and GABAR (GABAAreceptor(ɑ1-6)). Whole hippocampal tissue from the control and radiation groups was isolated and immediately sonicated in 200 μL of radio immunoprecipitation assay buffer (Beyotime Biotechnology, P0013B) containing 1× haltTMprotease and a phosphatase inhibitor single-use cocktail (Cat# 78443; Invitrogen,Waltham, MA, USA). The supernatant of the samples was collected after being centrifuged under 4°C at 15,364 ×gfor 15 minutes. Total protein quantification was performed using the PierceTMBCA Protein Assay Kit (Cat#23225, Invitrogen). Then, equal amounts (30 μg) of the protein samples were mixed with protein loading buffer, boiled, and analyzed via 10%polyacrylamide gel with 0.1% sodium dodecyl sulfate. The proteins were transferred to polyvinylidene fluoride membranes (0.2 μm, Merch Millipore,Carrigtwohill, Ireland), followed by 5% milk-blocking for 60 minutes at room temperature in Tris-buffered saline (TBS)/0.1% Tween-20, and consequently transferred in the presence of primary antibodies (see below) to TBS/0.1%Tween-20 overnight at 4°C. Following three 8-minute washes with TBS/0.1%Tween-20, the polyvinylidene fluoride membranes were incubated with goat anti-mouse secondary horseradish peroxidase-linked antibodies (1:5000,Cat# 7074S, RRID: AB_2099233, Cell Signaling Technology, Boston, MA, USA)or horse anti-rabbit secondary horseradish peroxidase-linked antibodies(1:5000, Car# 7076S, RRID: AB_330924, Cell Signaling Technology) for an hour at room temperature. Protein signals were visually identified through Clarity Western electrochemiluminescence substrate (Millipore, WBKLS0500)on a chemiluminescence apparatus (Tanon-5200C, Shanghai, China).Quantification was performed using ImageJ Fiji (https://imagej.net/software/fiji/). Primary antibodies used were: mouse antiα-tubulin (1:5000, Cat#66031-1, RRID: AB_11042766, Proteintech, Rosemont, IL, USA), rabbit anti-VGAT (1:1000, Cat# 131002, RRID: AB_887871, SySy), mouse anti-VGLUT1(1:1000, Cat# sc-377425, RRID: AB_2687960, Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse anti-GluR1 (1:1000, Cat# sc-55509, RRID: AB_629532,Santa Cruz Biotechnology), mouse anti-GluR2 (1:1000, Cat# sc-517265,Santa Cruz Biotechnology), mouse anti-NMDAɛ2 (1:1000, Cat# sc-365597,RRID: AB_10847218, Santa Cruz Biotechnology), and mouse anti-GABAA receptor(ɑ1-6) (1:1000, Cat# sc-376282, RRID: AB_10988210, Santa Cruz Biotechnology).
Statistical analysis
We employed GraphPad Prism 6.0 (GraphPad Software, San Diego, CA, USA)for statistical analyses. The experimental data were represented using dotblot figures, with error bars showing the mean ± standard error of mean(SEM). The observer was blinded to the experimental groups only for the electrophysiological data. Statistical significance was set atP< 0.05. We used independent samplet-tests for two-group comparisons. To assess betweengroup differences in escape latency in the training phase of the behavioral tests, and the differences in the number of spikes fired across a range of current injections, we used a two-way analysis of variance for repeated measures with Sidak’s multiple comparisons test.
Results
Radiation induces hippocampus-dependent memory deficits
To assess radiation-induced cognitive impairment, adult male mice that received a single 30 Gy dose of ionizing cerebral irradiation were subjected to tests of cognitive function at 3 months post-irradiation. These tests included the novel object location test and the MWM, which are both hippocampusdependent memory tasks (Figure 1A). In the novel object location test (Figure 1B), the radiation and control mice spent an equal amount of time exploring both objects on day 2 (Figure 1C). The control mice spent more time exploring the re-located object on day 3 (P< 0.0001), while the radiation mice spent an equal amount of time exploring the two objects (P= 0.335; Figure 1D).
For the MWM test, the mice were trained daily for 5 days to locate the hidden platform. The escape latency (the time taken to find the hidden platform) was longer in the radiation mice compared with the control mice on days 2 (P<0.001) and 3 (P< 0.001; Figure 1E), but there were no differences between these two groups on days 4 and 5. On day 6, the mice swam in the maze for 60 seconds during the probe test. The swimming patterns during the probe test are illustrated in Figure 1F. Compared with the radiation mice, the control mice spent a significantly different amount of time in the different quadrants (P< 0.0001), with the longest time spent in the target quadrant(Figure 1G). In contrast, the radiation mice spent a similar time amount oftime in all of the quadrants (P= 0.1979; Figure 1G). Moreover, the radiation mice spent significantly less time exploring the target quadrant (P= 0.0281;Figure 1G) and performed fewer crossings of the previous site of the hidden platform compared with those in the control group (P= 0.0051; Figure 1H).These data indicate that at three months post irradiation, the mice displayed a spatial memory deficit in the MWM test. We also conducted the cued test(visible platform) and found no significant between-groups difference in thetime taken to reach the visible platform (P= 0.3369; Figure 1I). Furthermore,the two groups had a similar mean swimming speed (Figure 1J). This suggests that the radiation did not affect visual or sensorimotor function in the mice.Taken together, these data indicate that a 30 Gy dose of radiation induced long-term spatial memory deficits in mice.
Radiation diminishes excitability of hippocampal CA1 pyramidal neurons
Given that the radiation mice showed hippocampal-dependent memory deficits, we used whole-cell patch clamp recording to assess whether cranial irradiation changed the intrinsic excitability of CA1 pyramidal neurons. CA1 pyramidal neurons from radiation mice exhibited a significant reduction in firing rate compared with those in the control mice (P= 0.0004;Figure 2A and B). We observed an increase in rheobase in the radiation group compared with the control group (P= 0.0005; Figure 2C), while the AP threshold (P= 0.3196; Figure 2D) and resting membrane potential (P=0.3197; Figure 2E) remained unchanged. We further examined alterations in neuronal excitability by calculating the AP latency and the amount of APs induced by depolarizing current ramps (steps from 0 to 160 pA within a 5-second duration) (Figure 2F). Similarly, we found that radiation decreased the total number of APs (P= 0.0006; Figure 2G) and increased the AP latency (P= 0.0003, Figure 2H), compared with the mice in the control group. These results indicate that radiation reduced the intrinsic excitability of CA1 pyramidal neurons.
Radiation reduces spontaneous excitatory transmission but increases spontaneous inhibitory transmission to hippocampal CA1 pyramidal neurons
To examine radiation-associated alterations in the excitatory input to hippocampal CA1 pyramidal neurons, we recorded AMPA receptor (AMPAR)-mediated sEPSCs (Vhold= -70 mV) (Figure 3A). The results showed that compared with the control group, the mean sEPSC frequency but not the amplitude in the radiation group was considerably reduced (frequency:P=0.018, amplitude:P= 0.7038; Figure 3B). We also recorded GABA receptormediated sIPSCs (Vhold= 0 mV) (Figure 3C) to enable a functional analysis of inhibitory synapses. We observed a significant enhancement in the mean frequency but not the amplitude of sIPSCs in the radiation group compared with the control group (frequency:P= 0.0302, amplitude:P= 0.7725; Figure 3D). All of these findings suggest that radiation reduced excitatory synaptic input and enhanced inhibitory input to hippocampal CA1 pyramidal neurons.
Radiation impairs hippocampal LTP
LTP is characterized by the persistent strengthening of synaptic activities,and has been associated with learning and memory (Titley et al., 2017).To determine the impact of radiation on synaptic plasticity, we recorded fEPSPs at Schaffer collaterals to CA1 synapses (Figure 4A). The input-output curves of the fEPSPs indicated that the radiation did not affect basal synaptic transmission (Figure 4B). We then recorded LTP for 60 minutes following one train of high-frequency stimulation (100 Hz with 50 pluses) (Figure 4C). In the radiation mice, we observed a significant descent in the fEPSP slope in the last 10 minutes compared with the control mice (P= 0.0284; Figure 4D).Our results indicate that exposure to cranial irradiation impairs LTP/synaptic plasticity in hippocampal CA1 neurons in mice.
Radiation induces molecular alterations in glutamatergic and GABAergic synapses in the hippocampus
Based on the above electrophysiological findings, we investigated the molecular mechanisms that contributed to the imbalance in excitatory and inhibitory synaptic input after radiation (Figure 5A). We first investigated the expression levels of VGLUT1 and VGAT in hippocampal CA1 neurons, which are regarded as excitatory and inhibitory presynaptic markers, respectively(Sando et al., 2017). Interestingly, immunofluorescence staining revealed a dramatic decline in VGLUT1-positive expression and an increase in VGATpositive expression in hippocampal CA1 neurons in both the pyramidal layer and stratum radiatum in the radiation versus control group (Figure 5B and C).We further confirmed similar molecular changes in VGLUT1 (P= 0.0485) and VGAT (P= 0.0054) in the hippocampus via western blot analysis (Figure 5DG). Western blot analysis of other neurotransmitter receptors that are vital to synaptic plasticity indicated a notable decrease in GluR1 expression (P=0.0434; Figure 5G and H), which is a subunit of AMPAR, but not in GluR2 (P= 0.7871; Figure 5G and I), NMDAɛ2 (P= 0.5459; Figure 5G and J) or GABAAreceptor(ɑ1-6) (P= 0.9738; Figure 5G and K) expression in the radiation group compared with the control group. In summary, our data indicated that the radiation mice exhibited significant alterations in synaptic markers of glutamatergic and GABAergic neurons in the hippocampus, providing mechanistic clues regarding the long-term effects of radiation with respect to neuronal dysfunction and cognitive impairment.
Discussion
Although cognitive deterioration after cranial irradiation is extensive and devastating, the mechanisms underlying the cognitive sequelae remain largely undetermined (Greene-Schloesser et al., 2013; Makale et al., 2017). Here,we identified changes in cognitive function, pathological characteristics,electrophysiological activity in hippocampal neurons, and associated molecular changes induced by a single 30 Gy dose of radiation in male mice. Our findings offer mechanistic insight regarding cognitive dysfunction following cranial irradiation, suggesting new avenues for therapeutic intervention.
The pathogenesis of irradiation-induced cognitive impairment depends on multiple factors, including the type of radiation used, radiation dose, and whether the treatment was single or fractionated (Bender, 2012; Boria and Perez-Torres, 2019; de Kruijff, 2020), as well as biological sources of variance such as genetic susceptibility (Wang et al., 2019) and sex-based differences(Hinkle et al., 2019; Boria and Perez-Torres, 2020). All of the findings in this study are based on male mice, which represents a limitation regarding the applicability of our data on the effects of cranial irradiation on the intrinsic excitability and synaptic plasticity to female mice. Although most previous animal studies of radiation-induced brain injury have used male animals,recent reports have demonstrated sex differences in the effects of cranial irradiation on cognitive dysfunction, brain necrosis, and spine loss (Hinkle et al., 2019; Boria and Perez-Torres, 2020). Thus, future studies are needed to investigate the influence of sex on the effects of radiation-induced brain injury.
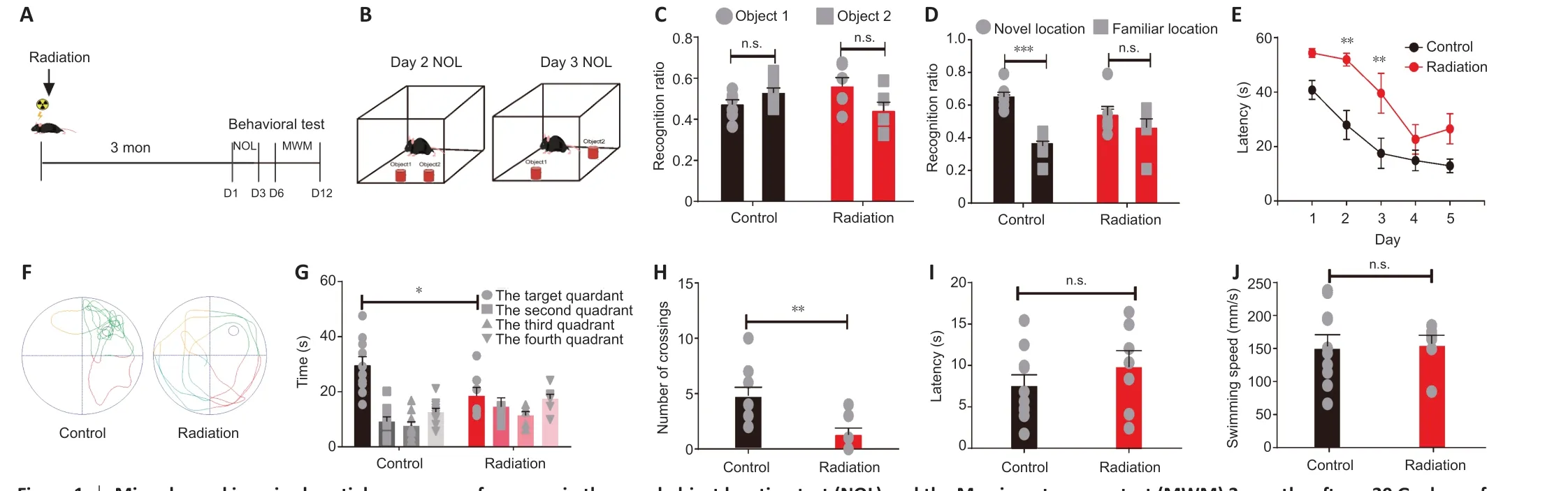
Figure 1 |Mice showed impaired spatial memory performance in the novel object location test (NOL) and the Morris water maze test (MWM) 3 months after a 30 Gy dose of radiation.(A) Timeline of the NOL and MWM tests. (B) Schematic representation of the NOL. (C) The control and radiation mice were not significantly different in terms of the recognition ratio between objects on day 2 (control: n = 7, independent sample t-test, P = 0.1222; radiation: n = 6, independent sample t-test, P = 0.0772). (D) The control mice had a higher recognition ratio for the relocated object on day 3, while this was not the case for the radiation mice (control: n = 7, independent sample t-test, P < 0.0001; radiation: n = 6,independent sample t-test, P = 0.3335). (E) The escape latency curve during the 5 training days (control/radiation: n = 10/7; two-way repeated measures analysis of variance: time: P <0.0001; group: P = 0.0009; Sidak’s multiple comparisons test: escape latency on day 2: P < 0.01, day 3: P < 0.01; other days: n.s.). (F) Illustration of the swimming pattern in the control and radiation mice during the probe test. (G) The time spent in each quadrant during the probe test (control/radiation: n = 10/7, unpaired Student’s t-test, P = 0.0281). (H) Number of crossings of the location of the hidden platform during the probe test (control/radiation: n =10/7, independent sample t-test, P = 0.0051). (I, J) The escape latency to the platform(I: control/radiation: n = 10/7; independent sample t-test, P = 0.3369) and the swimming velocity during the cued test (J: control/radiation: n = 10/7, independent sample t-test, P =0.8728). Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. n.s.: No significance.
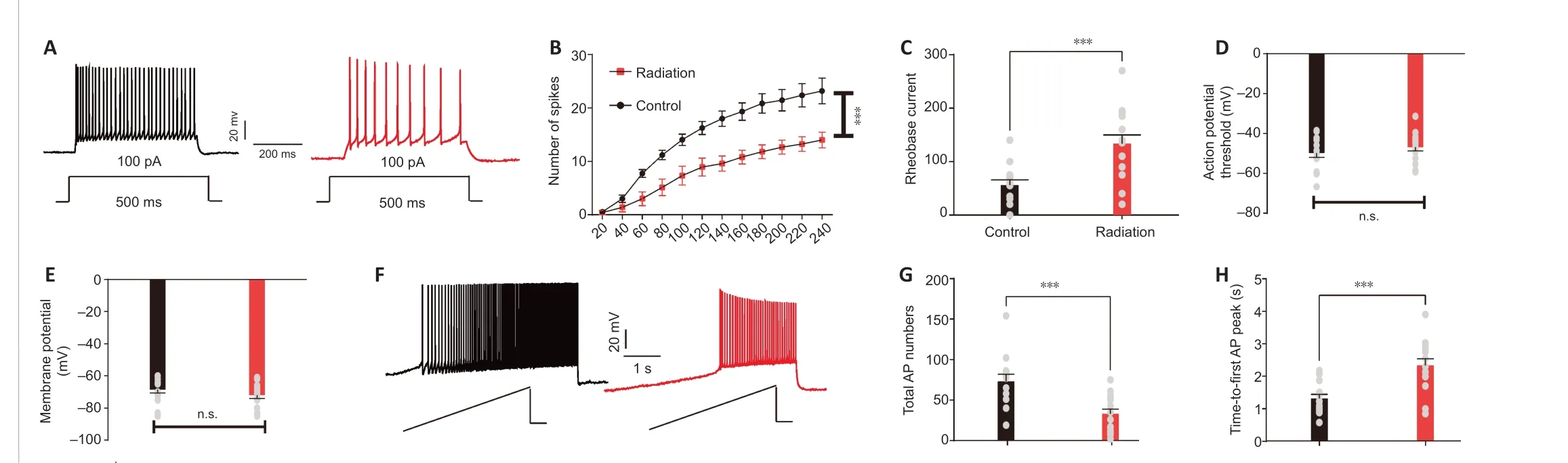
Figure 2 |Radiation diminished intrinsic excitability in hippocampal CA1 pyramidal neurons.(A) Representative firing patterns of hippocampal CA1 pyramidal neurons in the control (black) and irradiated (red) mice in response to depolarizing current injections (100 pA). (B)Averaging the number of spikes fired indicated a decrease in excitability after radiation across a range of current injections (control/radiation: n = 15/15, two-way repeated measures analysis of variance: current injection: P < 0.0001, group: P = 0.0004). (C) Increased rheobase current in the radiation mice (control/radiation: n = 14/15, independent sample t-test,P = 0.0005). (D, E) We found no significant difference in the action potential threshold (D: control/radiation: n = 14/15; independent sample t-test, P = 0.3196) or resting membrane potential (E: control/radiation: n = 14/15, independent sample t-test, P = 0.3197) between the groups. (F) Representative firing pattern of hippocampal CA1 pyramidal neurons from the control (black) and irradiated (red) mice in response to a series of depolarizing current ramps (steps from 0 to 160 pA with a 5-second duration). Scale bars: 20 mV (vertical axis),200 ms (horizontal axis). (G) The total number of APs was decreased in irradiated mice (control/radiation: n = 14/15, independent sample t-test, P = 0.0006). (H) Radiation delayed the time to the first AP peak (control/radiation: n = 14/15, independent sample t-test, P = 0.0003). Data are expressed as mean ± SEM. ***P < 0.001. AP: Action potential; n.s.: no significance.
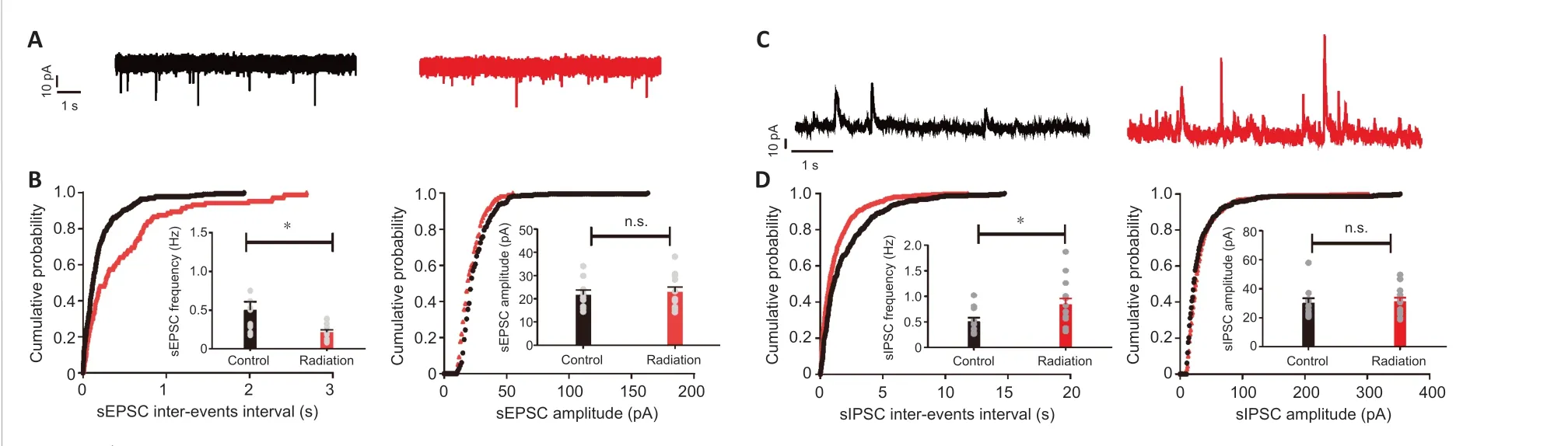
Figure 3 |Radiation decreased the sEPSC and increased the sIPSC in hippocampal CA1 pyramidal neurons.(A) Representative patterns of sEPSC traces in hippocampal CA1 pyramidal neurons from the control (black) and irradiated (red) mice. (B) Cumulative distribution plots and group data(insert) showed a significant decrease in the average frequency of sEPSCs (left) but not the average amplitude (right) in the irradiated mice (frequency: control/radiation, n = 10/12,independent sample t-test, P = 0.0118; amplitude: control/radiation: n = 10/12, independent sample t-test, P = 0.7038). (C) Representative patterns of sIPSC traces from hippocampal CA1 pyramidal neurons from the control (black) and irradiated (red) mice. (D) Cumulative distribution plots and group data (insert) showed a significant enhancement in the average frequency of sIPSCs (left) but not the average amplitude (right) in the irradiated mice (frequency: control/radiation: n = 11/15, independent sample t-test, P = 0.0302; amplitude:control/radiation: n = 11/15, independent sample t-test, P = 0.7725). Data are expressed as mean ± SEM. *P < 0.05. sEPSC: Spontaneous excitatory postsynaptic current; sEPSC:spontaneous excitatory postsynaptic current.
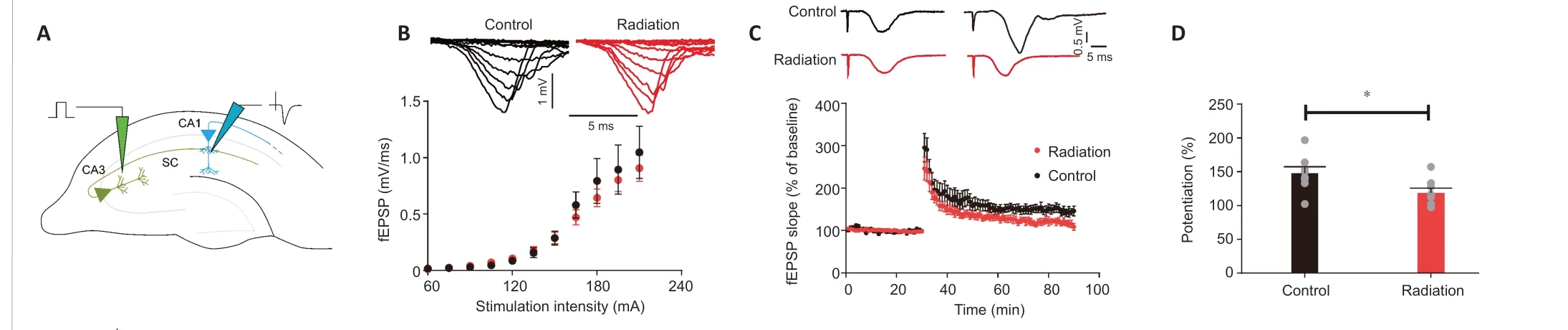
Figure 4 |Impairment of hippocampal LTP in the irradiated mice.(A) Schematic representation of LTP stimulation and recording in hippocampal CA1 and CA3 synapses. (B) Input-output curve of fEPSP slope (n = 8 slices/4 mice in each group). (C)Representative EPSP traces before and 1 hour after high frequency stimulation (100 Hz for 50 pluses) in the control and irradiated mice (upper), the average fEPSP plotted againsttime in minutes (lower) (n = 4 mice/group). (D) The average fEPSP slopes over the last 10 minutes of recording, normalized to baseline (control/radiation: n = 8/8, independent sample t-test, P = 0.0284). Data are expressed as mean ± SEM. *P < 0.05. EPSP: Excitatory postsynaptic potentials; fEPSPs: field excitatory postsynaptic potentials; LTP: long-term potentiation.

Figure 5 |Changes in glutamatergic and GABAergic synapses in the hippocampal CA1 of irradiated mice.(A) Timeline of western blot and immunofluorescence analysis. (B, C) Radiation induced a decline in VGLUT1 (Alexa Fluor 594 for red) but an increase in VGAT (Alexa Fluor 488 for green) expression in both the pyramidal layer (B) and stratum radiatum (C) of the hippocampal CA1. Scale bars: 10 μm. (D) The expression levels of VGLUT1 increased but those of VGAT decreased in the hippocampus of irradiated mice, as per western blotting, compared with the control mice. (E, F) Quantitative results of VGLUT1 and VGAT expression levels in the hippocampus of control and irradiated mice (control/radiation: n = 3/3, VGLUT1: P = 0.0485, VGAT: P = 0.0054). (G) The expression levels of GluR1, GluR2, NMDAɛ2, and GABAARɑ(1-6) in the hippocampus were evaluated in the control and irradiated mice by western blotting. (H-K) Quantitative results for GluR1, GluR2, NMDAɛ2, and GABAARɑ(1-6)expression levels in the hippocampus for the control and irradiated mice (control/radiation: n =3/3, GluR1: P = 0.0434, GluR2: P = 0.7871, NMDAɛ2: P = 0.5459, GABAARɑ(1-6): P =0.9738). Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01 (independent sample t-test). GABAAR: γ-Aminobutyrate type A receptor; NMDA: N-methyl-D-aspartic acid; VGAT:vesicular γ-aminobutyrate type A transporter; VGLUT1: type I vesicular glutamate transporter.
In the present study, we used a single instance of irradiation with a high dose in our animal model, as per previous reports (Xu et al., 2015). In animal studies and clinical practice, both single instances of irradiation with a high dose and fractionated irradiation with low dose are used (Yang et al.,2017; Milano et al., 2021). We previously reviewed and summarized the pathophysiological responses to radiation-induced brain injury in different animal models (Yang et al., 2017). Generally, fractionated irradiation carries a reduced risk of developing brain injury in comparison to a single high dose of irradiation. For example, a high cumulative dose (40 Gy) delivered via a fractionated irradiation model did not lead to vascular injury or demyelination at 6 weeks post irradiation (Semmler et al., 2013), and no cognitive deficits persisted after 7 months post irradiation (Lee et al., 2012). However, cranial irradiation delivered in a single high dose has several advantages for studying radiation-induced brain injury, including reproducible and stable phenotypes such as long-term cognitive impairment, vascular damage, white matter changes, and glial activation, which occur within weeks post irradiation(Hodges et al., 1998; Liu et al., 2010).
The type of radiation used is also vital to the pathogenesis of radiation-induced cognitive impairment. Linear energy transfer (LET; expressed as keV/μm)is used to describe the amount of energy deposited per unit of length when radiation passes through a material. High-LET radiation induces more damage per absorbed dose than low-LET radiation. Both high-LET radiation and low-LET radiation can uniquely affect neuroinflammation, neurogenesis, and neuronal morphology in animal models (Manda et al., 2009; Cacao and Cucinotta, 2019; Roobol et al., 2020). For example, altered neurogenesis at the early stage post-irradiation in animal models has been found to vary according to the type or dose of irradiation received (Manda et al., 2009;Rivera et al., 2013; Zanni et al., 2018).
To determine how the hippocampus contributes to radiation-induced cognitive deficits, we examined LTP in the CA1 hippocampal region. In contrast to a previous report on LTP in the dentate gyrus (Wu et al., 2012),we found no detectable change in the Schaffer collateral pathway of the hippocampus during the induction phase of LTP. However, we did find impaired expression of LTP. Importantly, we found that radiation induced a decrease in GluR1 expression in the hippocampus. The AMPAR plays a key role in synaptic plasticity (Malinow and Malenka, 2002; Shepherd and Huganir, 2007), and LTP of synaptic strength is reflected through the synaptic insertion of AMPARs, resulting in synaptic strength enhancement and an increase in spine size (Kopec et al., 2007). Since most of the recruited AMPARs have extrasynaptic origins during LTP formation (Malinow and Malenka, 2002;Patterson et al., 2010), the lack of functional AMPARs on the extrasynaptic surface of GluR1-deficient neurons will result in a major impairment of LTP(Zamanillo et al., 1999; Andrásfalvy et al., 2003; Granger et al., 2013). The radiation-induced loss of GluR1 that we observed in the hippocampus may have led to decreased extrasynaptic AMPARs and weakened synaptic strength,thus further contributing to the functional impairment of LTP expression. One recent report also found a loss of GluR1 expression after radiation (Krishnan et al., 2021), while another showed no detectable changes in GluR1 expression at 12 months post irradiation (Shi et al., 2006), indicating that age and/or time may contribute to the alteration of GluR1 expression after radiation.Radiation-induced impairments in LTP in other hippocampal pathways have also been reported (Zhang et al., 2018). Together with our findings, these data suggest that radiation may have a direct impact on mature neurons and their function in the hippocampus, partly due to altered GluR1 expression.
GluR1-induced synaptic potentials are the principal contributor to dendritic and somatic depolarization that drives APs (Nicholson and Geinisman,2009). We found that radiation triggered a prominent decrease in the depolarization-evoked firing activity of CA1 pyramidal neurons compared with non-irradiated neurons. Moreover, the effects of radiation on the intrinsic membrane properties of hippocampal neurons persisted three months post irradiation, resulting in an increased rheobase. A similar alteration in intrinsic electrophysiological properties was found to result in decreased excitability of CA1 pyramidal neurons (Tolliver and Pellmar, 1987; Sokolova et al., 2015),which is likely to affect microcircuit activity in the hippocampus. Moreover,recent reports have shown that radiation altered intrinsic properties of principal cells in the perirhinal cortex (Allen et al., 2020). These results further demonstrate that radiation can directly affect neuronal function.
A balance between excitatory glutamatergic and inhibitory GABAergic transmission in CA1 neurons is indispensable for the proper function of neuronal networks and for normal cognition. We found that exposure to radiation produced a decrease in the frequency of sEPSC in CA1 pyramidal neurons, suggesting that presynaptic glutamate release had decreased. This finding is in agreement with a previous study reporting that radiation led to greater presynaptic versus postsynaptic damage, and that the effect was dose and dose rate dependent and postsynaptic damage required a larger dose of radiation, and was not susceptible to dose rate (Tolliver and Pellmar,1987; Pellmar et al., 1990). In the present study, we found reduced levels of the presynaptic glutamatergic marker VGLUT1 in the CA1 pyramidal neurons of irradiated mice. Many studies have focused on changes in excitatory synapses after radiation. For instance, an increase in excitatory synapses was observed at the early stage after radiation (< 24 hours) (Duman et al., 2018).However, this acute increase had converted into synapse loss at later time points (> 90 hours) (Duman et al., 2018), and the decrease in spines lasted from days to weeks (Chakraborti et al., 2012; Parihar and Limoli, 2013).These results indicate that radiation ultimately causes an irreversible loss of excitatory synapses, leading to cognitive deficits. Future research is needed to investigate the precise mechanisms driving the conversion from acute increases in excitatory synapses to the loss of excitatory synapses in irradiated brains, and the associated consequences for cognition.
We observed an increase in inhibitory input to CA1 pyramidal neurons in the radiation mice, suggesting that exposure to radiation may increase the probability of GABA release from the synaptic terminal of inhibitory neurons.This result was further confirmed by the increased expression of the inhibitory presynaptic maker VGAT in the irradiated mice. GABA neurotransmission plays a key role in learning/memory processes by modulating synaptic plasticity(Olpe et al., 1993; Huang et al., 2005; Gong et al., 2009), neural oscillations(Gong et al., 2009; Mann and Mody, 2010), and neurogenesis (Ge et al., 2007;Pontes et al., 2013) in the hippocampus. Both GABA-mediated hyperfunction(Clarkson et al., 2010; Zurek et al., 2014; Lissemore et al., 2018; Schulz et al., 2019) and hypofunction (Gill et al., 2011; Han et al., 2014; ) can be the primary cause of cognitive impairment in pathological states such as ischemic stroke. Here, we observed an imbalance in the excitatory and inhibitory inputs to CA1 neurons, with significantly increased inhibition. Interestingly, enhanced GABA neurotransmission was also reported in the acute phase (30 minutes)post irradiation (Duman et al., 2018), as well as in 30-day-old irradiated juvenile mice (Caceres et al., 2013). Our findings therefore suggest that reducing GABA-mediated inhibitory neurotransmission in the hippocampus may have therapeutic potential for patients with radiation-induced cognitive impairment. Indeed, this strategy has been effective in treating other neurological diseases (Clarkson et al., 2010; Zurek et al., 2014; Schulz et al.,2019). However, a recent study reported significantly decreased levels of GABA and GABAAreceptors in the hypothalamus after radiation (Franco-Pérez et al., 2020). These studies suggest that the distinct responses to radiation in different brain areas may relate to variations in GABA signaling. There are several types of GABAergic interneurons in the hippocampus. Whether or not there exist cell type-specific interneurons that are sensitive to radiation remains unknown, as we did not observe changes in the expression of GABAAR expression in the hippocampus of irradiated mice. Additional investigations regarding the molecular mechanisms underlying the enhancement of GABA release from presynaptic terminals are warranted. This study had several limitations. First, we did not examine the molecular mechanisms that modulate the imbalance of excitatory-inhibitory hippocampal neuronal input after cerebral irradiation. Second, we did not examine the mechanisms by which reducing excessive GABA-mediated inhibition by GABAAreceptor antagonists may have therapeutic potential for the treatment of radiationinduced impairments in synaptic plasticity and cognitive function (Fernandez et al., 2007).
In conclusion, we characterized radiation-induced functional alternations in hippocampal neurons, as reflected by changes in intrinsic electrophysiology,synaptic plasticity, and molecular markers. These changes appeared as a long-term consequence of cranial irradiation. Our findings have important implications for understanding the etiology of radiation-induced cognitive impairment and the development of therapeutic strategies for treatment.
Author contributions:Study design: MYW, WJZ, YT, WJL; experiment implementation: MYW, WJZ, YY, SJL; data analysis: MYW, WJZ, JX, SQC;manuscript draft: MYW, WJZ, YT, WJL, PY, QL. All authors read and approved the final manuscript.
Conflicts of interest:There are no conflicts of interest.Editor note: YT is an Editorial Board member of Neural Regeneration Research.He was blinded from reviewing or making decisions on the manuscript. The article was subject to the journal’s standard procedures, with peer review handled independently of this Editorial Board member and their research groups.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Functional in vivo assessment of stem cell-secreted prooligodendroglial factors
- iGluR expression in the hippocampal formation, entorhinal cortex,and superior temporal gyrus in Alzheimer’s disease
- Exploiting Caenorhabditis elegans to discover human gut microbiotamediated intervention strategies in protein conformational diseases
- N-methyl-D-aspartate receptor functions altered by neuronal PTP1B activation in Alzheimer’s disease and schizophrenia models
- Aminopeptidase A and dipeptidyl peptidase 4: a pathogenic duo in Alzheimer’s disease?
- Ubiquitin homeostasis disruption,a common cause of proteostasis collapse in amyotrophic lateral sclerosis?