
Brain-derived cell-free DNA
2022-03-09 07:31DeanSouthwoodSanyuktaSinghZacChatterton
中国神经再生研究(英文版) 2022年10期
Dean Southwood, Sanyukta Singh, Zac Chatterton
Following cell death, DNA can be released into the blood plasma and other body fluids in the form of cell-free DNA (cfDNA).These DNA fragments are typically ~167 bp in length, corresponding to the length of DNA wrapped around one nucleosome core (147 bp), plus DNA tails that survive endogenous DNase digestion (~10 bp).CfDNA is derived from a variety of sources,each having unique diagnostic applications.During pregnancy, fetal-derived cfDNA within an expectant mother’s blood plasma can diagnose fetal genetic abnormalities and is a well-established method for non-invasive prenatal testing. Cancer-derived cfDNA is also detectable within blood plasma by high sensitivity methods (e.g., NGS, ddPCR)and can accurately diagnose cancers by a minimally invasive blood test (Diehl et al.,2008). The short half-life of cfDNA (< 2 hours)also facilitates real-time measurement of disease burden, such as monitoring cancer relapse. There is now growing evidence of brain-derived cfDNA (bd-cfDNA) within the cerebrospinal fluid (CSF) and blood plasma.In this perspective, we review the present understanding, current challenges, and potential utility of bd-cfDNA.
The current state of bd-cfDNA: The cell-oforigin of CSF or blood plasma bd-cfDNA may be identified using brain-specific genetic and epigenetic markers (Figure 1). Targeted deep sequencing of blood plasma cfDNA for genes known to harbor mutations (e.g., EGFR) can detect bd-cfDNA derived from neurological cancers. For example, genetic mutations are detectable in ~50% of glioblastoma multiforme patients. Notably, this prevalence is below that seen in non-neurological cancers (typically ~85% of patients), likely due to a lower tumor fraction (< 0.5% for glioblastoma multiformevs. > 1% for nonneurological cancers) (Zill et al., 2018). In non-malignant brain diseases, somatic mutations can also be used to detect bdcfDNA within the CSF. For example, we recently identified brain-specific somatic mutations in TSC1 in the CSF of a patient with focal cortical dysplasia (Ye et al., 2021).The variant allele frequency (analogous to tumor fraction) of this somatic mutation was 7.8%, indicating that CSF contains a higher proportion of bd-cfDNA than blood plasma,whereas the total amount of cfDNA in blood plasma is ~20 times higher than in CSF (Pan et al., 2020).
In the absence of genetic markers, bdcfDNA may be detected via epigenetic markers, namely patterns of brain-specific DNA methylation covalently bound to cfDNA fragments. In a seminal study,Lehmann-Werman and colleagues used bisulfite sequencing, targeting genomic regions known to harbor brain-specific DNA methylation, to identify bd-cfDNA within the blood plasma of patients following traumatic brain injury, ischemic brain damage following cardiac arrest, and multiple sclerosis (Lehmann-Werman et al., 2016).Most multiple sclerosis patients had > 0.2%detectable bd-cfDNA, which was highest during relapse and likely derived from oligodendrocytes. Marked increases in bdcfDNA were observed in acute (< 24 hours)sampling following ischemic brain damage,in which most patients had > 4% bd-cfDNA;however, the day-to-day variability was also notably high. Curiously, the highest amounts of bd-cfDNA in traumatic brain injury were not necessarily detected in the acute stages and occasionally peaked more than 1 week post-injury. We recently observed significant increases in the blood plasma levels of neuron-derived cfDNA in subjects less than 2.5 hours after mild trauma; however, these increases were not consistently accompanied by increases in glia-derived cfDNA levels(Chatterton et al., 2021). The variability in the timing and origin of bd-cfDNA may have important clinical implications similar to neurological proteins; for example, increased blood serum levels of the glial proteinGFAPcan prognosticate traumatic intracranial abnormalities and guide clinical decision making, such as the need for a CT scan or radiation exposure (Bazarian et al., 2018).
New genetic targets for bd-cfDNA analysis:Non-malignant mosaic somatic mutations are postzygotic genomic changes which result in distinct cell populations, with different genotypes, within the same organism. Somatic mosaicism in postmitotic neurons will largely persist for the life of the individual, whilst mosaic somatic mutations in multipotent neural precursor cells give rise to neuronal and glial populations with distinct genotypes. Somatic mutations are present in almost all individuals to a small degree, on the order of 5-20 mutations within proteincoding genes (Kim et al., 2021); notably,however, they have been identified in patients with cerebral cortical malformations(e.g.,DCXin double-cortex syndrome),neuropsychiatric and neurodevelopmental disorders, and neurodegenerative disease(Nishioka et al., 2019) (Figure 1). At least some somatic mutations are disease-causing- deleterious mutations have been found to affect disease-associated genes in epilepsy and autism spectrum disorder, for example(Stosser et al., 2018; Rodin et al., 2021).Characterizing mosaic somatic mutations in neurological diseases has the potential to further elucidate disease mechanisms whilst advancing disease subtyping.However, research is hindered in part by the high depth of sequencing required to provide sufficient certainty of low-frequency mutations in blood, or the need for biopsied brain tissue, usually taken post-mortem. The analysis of bd-cfDNA within CSF or blood plasma provides a less invasive avenue by which to determine the burden of mosaic somatic mutations in the brain during life (Ye et al., 2021) (Figure 1).
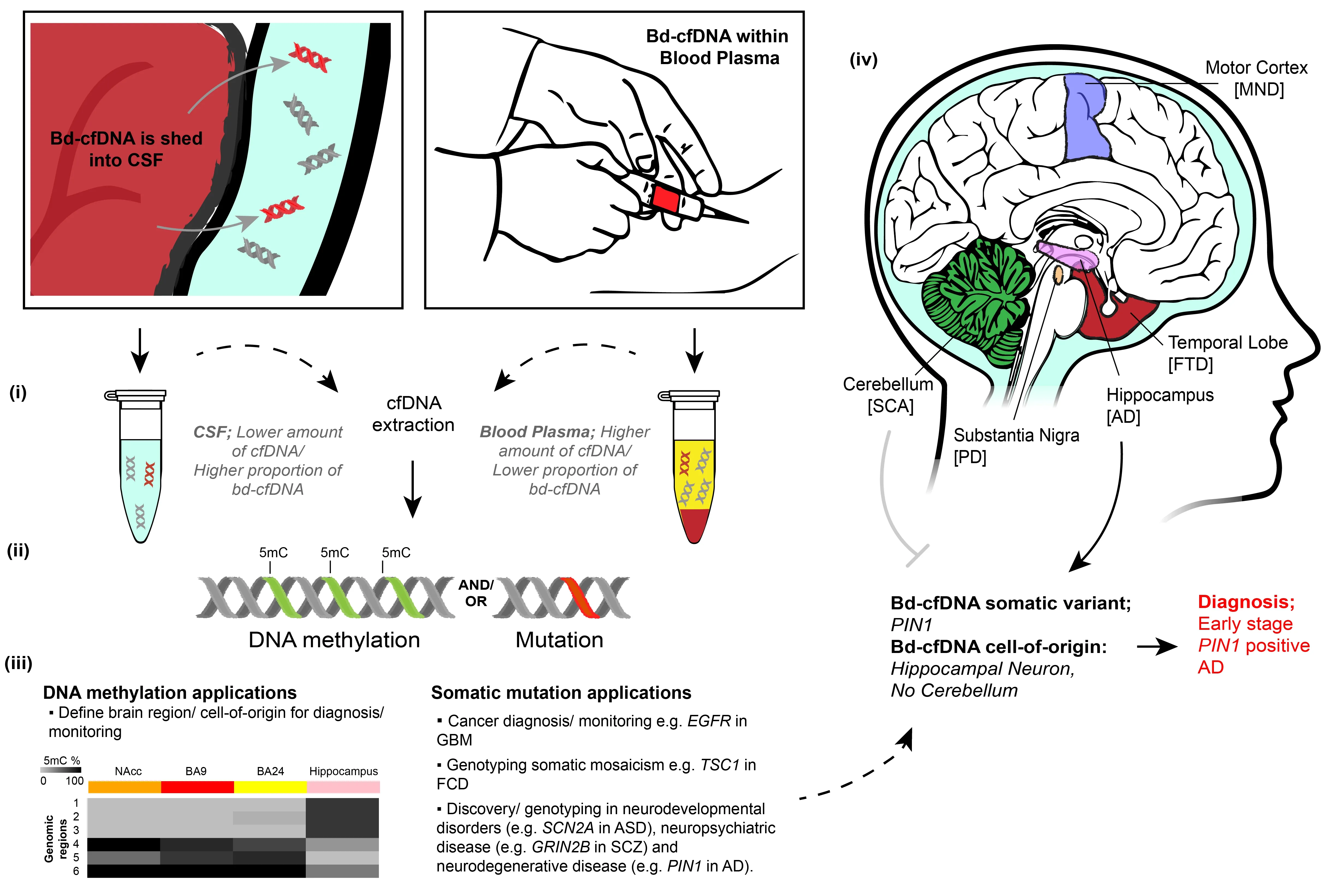
Figure 1 |Brain-derived cfDNA overview and potential applications.Bd-cfDNA is shed into the CSF (left box) and/or blood plasma (right box). (i) Following cfDNA extraction, the CSF contains a lower amount of cfDNA, but a higher proportion of bd-cfDNA, compared to the blood plasma. (ii) CfDNA retains both genetic and epigenetic information. (iii) Epigenetic information (DNA methylation) can define the cell-of-origin for diagnosis/monitoring, with example DNA methylation shown (left) between neurons of the hippocampus and BA24,BA9, and nucleus accumbens (NAcc) brain regions for three genomic regions across N2RF2 (1-3), two intragenic regions of KCNIP1 (4) and FGFR3 (6) and a non-genic region of chromosome 5 (5) recently discovered (Rizzardi et al., 2019).Genetic information may also be used to diagnose/monitor cancer and neurological disorders with somatic mosaicism(right). Combined, this information can identify that cfDNA is brain-derived and can genotype neurodevelopmental,neuropsychiatric, and neurodegenerative diseases. An example application for the combined genetic and epigenetic analysis of bd-cfDNA is shown in iv. Distinct brain regions are affected by neurodegenerative diseases. The detection of bd-cfDNA of the somatic variant PIN1 from hippocampal neurons but the absence of bd-cfDNA from the cerebellum leads to the diagnosis of early-stage PIN1 positive AD. AD: Alzheimer’s disease; ASD: autism spectrum disorder; FCD:focal cortical dysplasia; FTD: frontotemporal dementia; GBM: glioblastoma multiforme; MND: motor neuron disease;PD: Parkinson’s disease; SCA: spinocerebellar ataxia; SCZ: schizophrenia. Unpublished data.
New epigenetic targets for bd-cfDNA analysis: The genomic position of DNA methylation is highly organized and critical for the development of brain cell types,giving rise to a plethora of DNA methylation loci distinct between brain cell subtypes(Lister et al., 2013; Luo et al., 2017),brain regions (Rizzardi et al., 2019), and hemispheres (Li et al., 2020). These loci represent viable markers to determine the brain cell type of bd-cfDNA. Using these distinct DNA methylation patterns,we recently reported the first evidence of glia- and neuron-derived cfDNA within the blood plasma of subjects following mild trauma (Chatterton et al., 2021). The clinical symptoms that characterize neurological disorders arise from distinct brain regions or cell populations being affected. For example,the motor cortex is one of the predominant regions of the central nervous system affected by motor neuron disease, whereas the temporal lobe is predominantly affected in genetically related, but clinically distinct,frontotemporal dementia (Figure 1). During neurodegenerative disease progression,additional brain regions are often compromised; for example, the hippocampus is affected in the early stages of Alzheimer’s disease whilst the cerebellum is largely spared until later stages. DNA methylation profiles of brain cell types that identify the cell-of-origin of cfDNA fragments hold great potential for the diagnosis and monitoring of chronic neurodegenerative disease using bdcfDNA.
It is well established that cancers have distinct genome-wide DNA methylation profiles, which can provide many useful genomic sites to identify cancer-derived cfDNA (Chatterton et al., 2014). Indeed,DNA methylation analysis outperforms the analysis of canonical mutation sites in the diagnosis of cancer using cfDNA (Liu et al.,2020). In neurological disease however, while brain-cell-type-specific DNA methylation alterations are beginning to be described in cases such as schizophrenia (Mendizabal et al., 2019), they have yet to be investigated in genetic subtypes, germline or somatic.The assessment of DNA methylation in brain cell types has largely been hampered by technical limitations on brain cell type isolation. However, with the emergence of single-cell DNA methylation technologies,brain-cell-type-specific analyses are now possible and are likely to reveal a variety of distinct DNA methylation profiles that can be leveraged for neurological-disease-specific bd-cfDNA assays (Luo et al., 2017).
Neurological disorders account for 6.7% of total disability-adjusted life years (World Health Organization). With an aging global population, this burden is only expected to increase. As the brain is hard to biopsy,blood tests for neurological disorders would significantly advance molecular diagnostics in neurology and further our understanding of neurological disease mechanisms. There are still some challenges to be overcome,solutions to which would increase the utility of bd-cfDNA analysis more broadly.The relatively low abundance of bd-cfDNA(particularly in blood plasma) currently requires targeted analyses using high sensitivity techniques to detect, as well as tailored statistical and computational approaches to minimize false positive and false negative rates. In addition, while the short half-life of cfDNA is useful for realtime analysis, it is important to consider this variability when analyzing more longterm effects, where multiple sample times may be required. The future development of assays, targeting genomic regions affected by somatic mosaicism or harboring brainspecific DNA methylation, will improve the sensitivity and specificity of bd-cfDNA detection, and broaden the scope of neurological disorders that may benefit from bd-cfDNA analysis.
ZC is generously funded by The University of Sydney Postdoctoral Fellowship. ZC and DS are generously funded by a National Health and Medical Research Council(NHMRC)-European Union Joint Program on Neurodegenerative Disease Research Grant(JPND2019-466-261 & APP11912407).
Dean Southwood, Sanyukta Singh,Zac Chatterton*Brain and Mind Center, School of Medical Sciences,Department of Neuroscience, The University of Sydney, Camperdown, NSW, Australia
*Correspondence to: Zac Chatterton, PhD,zac.chatterton@sydney.edu.au.https://orcid.org/0000-0002-6683-1400(Zac Chatterton)
Date of submission: July 25, 2021
Date of decision: September 30, 2021
Date of acceptance: November 9, 2021
Date of web publication: February 28, 2022
https://doi.org/10.4103/1673-5374.335794
How to cite this article:Southwood D, Singh S,Chatterton Z (2022) Brain-derived cell-free DNA.Neural Regen Res 17(10):2213-2214.
Availability of data and materials:All datagenerated or analyzed during this study are included in this published article and itssupplementary information files.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix, tweak, and build upon the work non-commercially, as long asappropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Lee J Martin, Johns Hopkins University School of Medicine, USA.
Additional file:Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- Functional in vivo assessment of stem cell-secreted prooligodendroglial factors
- iGluR expression in the hippocampal formation, entorhinal cortex,and superior temporal gyrus in Alzheimer’s disease
- Exploiting Caenorhabditis elegans to discover human gut microbiotamediated intervention strategies in protein conformational diseases
- N-methyl-D-aspartate receptor functions altered by neuronal PTP1B activation in Alzheimer’s disease and schizophrenia models
- Aminopeptidase A and dipeptidyl peptidase 4: a pathogenic duo in Alzheimer’s disease?
- Ubiquitin homeostasis disruption,a common cause of proteostasis collapse in amyotrophic lateral sclerosis?