
Status of precision medicine approaches to traumatic brain injury
2022-03-09 07:22SahithiReddiSmitaThakkerVariaJanetAlderAnnaGiarratana
中国神经再生研究(英文版) 2022年10期
Sahithi Reddi, Smita Thakker-Varia, Janet Alder, Anna O. Giarratana
Abstract Traumatic brain injury (TBI) is a serious condition in which trauma to the head causes damage to the brain, leading to a disruption in brain function. This is a significant health issue worldwide, with around 69 million people suffering from TBI each year. Immediately following the trauma, damage occurs in the acute phase of injury that leads to the primary outcomes of the TBI. In the hours-to-days that follow, secondary damage can also occur, leading to chronic outcomes. TBIs can range in severity from mild to severe, and can be complicated by the fact that some individuals sustain multiple TBIs,a risk factor for worse long-term outcomes. Although our knowledge about the pathophysiology of TBI has increased in recent years, unfortunately this has not been translated into effective clinical therapies. The U.S. Food and Drug Administration has yet to approve any drugs for the treatment of TBI; current clinical treatment guidelines merely offer supportive care. Outcomes between individuals greatly vary, which makes the treatment for TBI so challenging. A blow of similar force can have only mild, primary outcomes in one individual and yet cause severe, chronic outcomes in another.One of the reasons that have been proposed for this differential response to TBI is the underlying genetic differences across the population. Due to this, many researchers have begun to investigate the possibility of using precision medicine techniques to address TBI treatment. In this review, we will discuss the research detailing the identification of genetic risk factors for worse outcomes after TBI,and the work investigating personalized treatments for these higher-risk individuals. We highlight the need for further research into the identification of higher-risk individuals and the development of personalized therapies for TBI.
Key Words: apolipoprotein E; biomarkers; brain injury; brain-derived neurotrophic factor; clinical trials; personalized treatments; precision medicine; tau; translational research; traumatic brain injury
Introduction
Traumatic brain injury (TBI) is a serious condition in which head trauma causes brain damage, leading to a disruption in brain function (Menon et al., 2010).TBI is a heterogeneous disease, and there are numerous causes, severities,and pathophysiologies of the injury that interface with personal genetic factors, complicating efforts to develop effective therapies. TBI is a significant issue, with a global incidence of 69 million and a global economic burden of$400 billion USD each year (Dewan et al., 2018). Broadly, TBIs are classified into three subtypes of mild, moderate, and severe by the Glasgow Coma Scale (GCS). By far, the most common type of TBI is a mild TBI, which occurs nearly 10 times that of both moderate and severe TBIs. Mild TBI can be quite disabling, with many patients reporting symptoms persisting months or years post injury (Dewan et al., 2018). Currently, there is limited understanding as to which patients will improve back to baseline and which will develop longterm complications. In order to develop better diagnostic, prognostic, and therapeutic tools, research is being done into the “silent factors” that may cause worse long-term outcomes after TBI.
This review will begin with a broad introduction to the problems associated with TBI. We will discuss the way in which TBI is currently clinically handled,from patient symptomatology to neuroimaging methods. We will highlight the shortcomings in our current ability to effectively diagnose and treat TBI patients, discuss the pathophysiology that is underlying the secondary diseases processes, and highlight biomarkers that show promise for clinical use. We will then go into detail about the role that genetic polymorphisms and other factors play on differential outcomes seen in TBI, and the targeted treatment approaches that are being taken to develop better therapeutics in both preclinical and clinical stages. We will close with a discussion of future directions, including the potential for development of a polygenic risk score for TBI.
Currently, the GCS is the most widely used tool to diagnose TBI severity. It is a simple diagnostic tool with a 3-15 rating scale, whereby a mild TBI is scored between 13-15, a moderate TBI is scored between 9-12, and a severe TBI is scored between 3-8. The GCS is used for both clinical and research purposes,however it has a number of limiting factors. For example, patients with severe TBI often have confounding factors which include endotracheal intubation and/or medical sedation-paralysis, which can result in over-estimation of neurological severity (Stocchetti et al., 2004).
To determine the neuropathology of injuries and diagnose TBI, neuroimaging may be used. In particular, computed tomography (CT) scans are commonly done in an emergency context after injury. In order to incorporate these findings into the greater clinical picture, scales have been developed to take the neuroimaging findings and improve prognostics. The most commonly used scale is the Marshall Scale, which uses CT scan findings to classify TBIs into six different categories (Marshall et al., 1991). However, the Marshall scale does not include a number of variables, so the Rotterdam scale was created (Maas et al., 2005). Both of these scales have shown utility in predicting the risk of increased intracranial pressure in the subacute time period after moderate and severe TBI. However, both have failed to predict long-term functional outcomes, indicating the need for further development of neuroimaging tools(Frodsham et al., 2020).
In order to improve clinical understanding of the heterogeneous long-term outcomes after TBI, the mechanisms of injury must be elucidated. Currently,the molecular mechanisms that contribute to long-term complications are not well understood. The effects of TBI occur in two phases, the acute primary injury process, followed by secondary injury (Menon et al., 2010). The primary injury is induced by the direct tissue damage caused at the time of the TBI.Injuries can be caused by direct impact blows, penetrating injuries that breach the skull, blast waves from explosions, and either rapid acceleration or deceleration. The acute damage that occurs can be heterogeneous in nature, and includes contusions, hematomas, hemorrhages, the shearing of white matter tracts commonly referred to as diffuse axonal injury, and cerebral edema (Moen et al., 2012). The secondary injury process is caused by numerous molecular mechanisms, starting from the time of injury and extending out for hours or days, resulting in neuronal cell death, axonal injury,inflammation, and neurodegeneration. Specifically, glutamate excitotoxicity,cell-mediated apoptosis, activation of neuroinflammatory processes, gliosis,cell body injury, tauopathy, dendritic injury, demyelination, necrosis, autoimmunity, and mitochondrial dysfunction all can occur in this secondary brain injury phase (Laskowitz et al., 2010). While these processes have been targeted in preclinical work for the development of therapeutics, there have been no successful clinical trials where treatment of these pathologies has shown a clear benefit to patients.
Given this obvious need for improved therapeutics to treat TBI, the medical and scientific communities have been pushing for the development of precision medicine approaches to treatment. Precision medicine uses what we know about subgroups of patients, for example their genetic profile, in order to develop a treatment method that is targeted to specifically to them (König et al., 2017). The hope of using precision medicine is that we can create more efficacious treatments with fewer side effects. In this review, we will discuss three genetic risk factors in particular that show promise as targets for precision medicine treatment approaches. The three genetic risk factors that we have identified are genetic polymorphisms in apolipoprotein E (APOE), brain-derived neurotrophic factor (BDNF), and Tau. They were selected for this review due to the promising studies using these risk factors and their interrelationship with each other.
Search Strategy
Searches of PubMed between February 2021 and April 2021 were conducted using the following keywords: “brain injury” + “polymorphism” OR “precision medicine” OR “personalized medicine” OR “personalized therapeutics”OR “polygenic risk score” OR “behavioral outcomes” OR “neurobiological outcomes” OR “targeted treatment.” Searches of the clinical trials database were conducted using the following keywords: “traumatic brain injury” OR“apolipoprotein E” OR “brain-derived neurotrophic factor” OR “tau.” Relevant papers were included in this narrative review, with the majority of papers being published within the last 5 years and less than 30% being published before 5 years.
Biomarkers
To develop better diagnostics and prognostics for patients, researchers began to identify biomarkers for clinical care. Biomarkers have the potential to determine the level of injury due to a specific mechanism (Figure 1). For example, if there is damage to astrocytes, glial proteins such as glial fibrillary acidic protein (GFAP) and S100β, a CNS-specific astrocyte calcium binding protein, may be elevated in plasma after injury. In separate studies, it was found that patients with elevated levels of plasma S100β had worse GCS scores 6 months post injury (Goyal et al., 2013) and elevated levels of GFAP were associated with incomplete patient recoveries. On the other hand,increased levels of neuronal-specific proteins such as ubiquitin C-terminal hydrolase-L1 (UCH-L1) in plasma may indicate there has been neuronal cell damage. One study detected a strong negative correlation between outcomes and UCH-L1 in the plasma in the first three days following injury, suggesting elevated levels of UCH-L1 are associated with worse outcomes (Takala et al.,2016). C-reactive protein (CRP) is a useful biomarker of general inflammation following TBI. A TBI pilot study showed that high sensitivity assays of CRP measured within 2 weeks of a TBI may be a good prognostic biomarker for disability 6 months later as elevated plasma levels are associated with poor outcomes (Xu et al., 2021).
In TBI cases where there is quick and complete resolution of symptoms,there is often a robust neural regeneration process occurring. Brain-derived neurotrophic factor (BDNF) is a neurotrophin that plays a pivotal role in synaptic plasticity, neuronal survival and regeneration. In a 2016 pilot study,altered serum BDNF levels on the day of injury were shown to be associated with a TBI diagnosis. Median BDNF concentrations were lower among the TBI cases relative to the uninjured controls and levels were higher in mild than in moderate or severe TBI. Thus, subjects that had lower BDNF values had increased chance of an incomplete recovery than those who had higher BDNF values (Korley et al., 2016). If there is injury to microtubules, levels of total (T-Tau) and/or phosphorylated tau (P-Tau) may be used as a biomarker following TBI (Wang et al., 2018). Tau is a protein that stabilizes microtubules in axons and has been implicated as a factor in chronic traumatic encephalopathy (CTE), a neurodegenerative condition linked to repeated TBIs.A 2016 multicenter prospective cohort study was done in the top professional Swedish hockey league. They found that T-Tau levels were elevated in the plasma and serum immediately following a TBI, and that levels decreased throughout the rehabilitation process (Shahim et al., 2014).
Although biomarkers have been shown to have potential as clinical diagnostics, they are not used in routine clinical place in the medical field today. However, researchers have developed an FDA-approved novel biomarker screen, the Banyan Trauma Indicator, to help streamline the decision for head CT scans in mild to moderate TBI. The Banyan Trauma Indicator measures levels of UCH-L1 and GFAP, as they are released from the brain into the bloodstream within 12 hours of injury and a negative assay result is associated with an absence of acute intracranial lesions, sparing the need for unnecessary scans (Su et al., 2019). While we discuss a few of the known biomarkers for TBI that are relevant here, there are many more that have been discussed in previous reviews (Wang et al., 2018). An increased understanding of additional biomarkers across the TBI spectrum is needed in order to develop further assays that can be used in clinical practice.
Genetic Risk Factors
TBI is a multifactorial disease that is complicated by the fact that each patient has a unique genetic background, which affects the pathophysiology of the disease process. While some patients sustain a TBI and recover within weeks, other patients may sustain a very similar TBI, and suffer from longterm consequences. One reason for this is the genetic variability within the world population. Researchers have begun to highlight certain genetic polymorphisms, which can contribute to a patient having worse outcomes after a TBI (Figure 2).
Apolipoprotein E
Produced by astrocytes, apolipoprotein E (ApoE) is a significant neuronal gene that is responsible for the transport and clearance of extracellular lipids and cholesterol within the brain. There are three main isoforms as a result of two single nucleotide polymorphism sites - apoe2, apoe3 and apoe4. ApoE3 is the most common human allele with an allelic frequency ranging from 53.6%to 89.8% and the ancestral ApoE4 allele has a frequency ranging from 5.2 to 40.7% (Corbo and Scacchi, 1999). As the human apoe4 protein is known to be functionally deficient compared to the apoe3 protein, many studies use humanizedAPOEmice as a clinically relevant model to study the effect ofAPOEgenetic polymorphisms on health outcomes. Specifically, these studies have shown thatAPOE4carriers have the largest known genetic risk factor for Alzheimer’s disease development and progression. Recently, there has been an interest in studying the effect of the ApoE4 allele on outcomes and recovery following various types of TBI.
To elucidate the effect ofAPOEpolymorphisms on recovery after TBI, several animal studies have been performed. Some rodent studies attribute the fact that the ApoE4 allele has worse outcomes to its role in blood-brain barrier(BBB) permeability and repair. In a controlled cortical impact (CCI) model of TBI, more severe neurological deficits were found in APOE4 injured mice than inAPOE3injured or wild type mice due to a significant sustained loss of tight junction proteins and enhanced matrix metallopeptidase-9 profiles, resulting in impaired BBB stabilization (Teng et al., 2017).APOE3andAPOE4mice had similar BBB responses, however,APOE3mice displayed faster spontaneous BBB repair thanAPOE4mice following TBI (Main et al., 2018). Another study found that after repetitive blast induced injury, ApoE4 injured mice had elevated levels of p-tau, which can result in neurofibrillary tangles that contribute to cognitive decline, relative to APOE3 injured mice (Li et al., 2017).Previous research in our lab studying the role of the E4 allele in a Lateral Fluid Percussion model of repeated mild TBI showed thatAPOE4injured transgenic mice have elevated inflammatory markers (TNF-a, CXCL1, IL-6) at 1 day post injury (DPI) and more edema at 21 DPI compared to APOE3 injured mice. In addition, we found that APOE4 injured mice have a higher occurrence of cell death and neurodegeneration at 1 DPI, indicating that the E4 allele is linked to more apoptosis in the early phase and more inflammation that continues into the later phase of recovery. Another significant finding from our lab was thatAPOE4injured mice had less total BDNF at 1 and 21 DPI, suggesting that differences in the BDNF pathway could cause other cellular and behavioral deficits following injury (Giarratana et al., 2020). Even with most animal research supporting ApoE4 as a risk allele, one conflicting study found that the ApoE4 genotype had no effect on outcomes following a repeated mild CCI model (Mannix et al., 2013).
Furthermore, a number of human pilot studies have been conducted to further investigate the role of theAPOE4allele on post-TBI outcomes,although many more are currently underway. One study found that the mean regional cerebral oxygen saturation ofAPOE4carriers was lower than that of non-APOE4carriers, suggesting increased risk of cerebral ischemia and hypoxia following TBI (Wu et al., 2020). A human study with 93 patients who were admitted to a neurosurgical unit post-TBI showed that 57% of patients with the APOE4 allele had an unfavorable outcome compared to 27% of patients without theAPOE4allele.APOE4patients were more than twice as likely as those withoutAPOE4to have unfavorable outcomes 6 months after head injury (Teasdale et al., 1997). In addition, patients with the APOE4 allele were found to have a slower recovery rate than those without the E4 allele (Alexander et al., 2007). While many studies show significant deficits with the E4 allele, others show no clear APOE genotype influence on neuropsychological outcome in mild and moderate TBI patients at 6 weeks and 6 months post injury (Shadli et al., 2011).
Many animal and human studies suggest that the E4 allele is associated with worse outcomes relative to E3 allele due to various signaling pathways and mechanisms such as those that involve histone deacetylases. It has been found that APOE4 increases nuclear translation of histone deacetylases,reducing levels of beneficial neurotrophic factors such as BDNF in the cell (Sen et al., 2015). Thus, these findings underscore the importance of considering genetic risk factors when evaluating neurotrauma patients and treating cognitive deficits through potential personalized therapies.
Tau
Previously mentioned as a potential biomarker for neurodegenerative processes after TBI, tau is an important protein in the brain that stabilizes microtubules in axons. When tau becomes phosphorylated at many sites(hyperphosphorylated) this can lead to the formation of neurofibrillary tangles, which have been found in the brains of people affected by the neurodegenerative brain process that is often a consequence of a history of multiple TBIs and CTE (Smith et al., 2013). Because of its important role in brain equilibrium, genetic polymorphisms in tau could alter its function,and thereby modify how the brain is able to heal after TBI. However,there have only been a few studies that have examined the role of genetic polymorphisms in tau and TBI. An early study investigated the association ofAPOE,APOEpromoter, and tau polymorphisms on the risk of sustaining a concussion in an athlete population, finding thatAPOEpromoter G-219T TT and the tau 53 genetic polymorphisms provide a risk factor for concussion history (Terrell et al., 2008). More recently, a 2019 study investigated the rs2435211 and rs2435200 single nucleotide polymorphisms (SNPs), and found that in their athlete population, the tau rs2435200 AG genotype might be associated with an increased susceptibility of sustaining multiple concussions(Abrahams et al., 2019). However, whether genetic polymorphisms in tau are responsible for worse outcomes after TBI remains to be investigated. A 2020 study found that genetic polymorphisms inAPOEmay be responsible for increased risk of neurodegeneration after TBI, withAPOE4carriers having higher tau burden (Vasilevskaya et al., 2020). Further studies are warranted to determine if there are genetic risk factors in tau which may contribute to poor outcomes after TBI, especially in regard to the neurodegenerative sequelae seen.
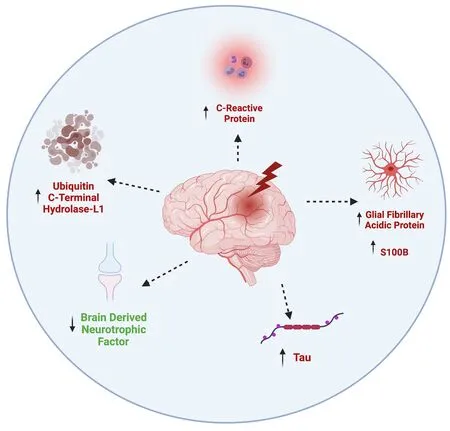
Figure 1 | Effects of TBI on biomarker levels.TBI is associated with increased levels of C-reactive protein levels, indicating inflammation, increased glial fibrillary acidic protein and S100B levels in astrocytes,increased phosphorylated tau and increased ubiquitin C-terminal hydrolase-L1 levels,indicating apoptosis. TBI is also linked to lower levels of brain derived neurotrophic factor, leading to poor recoveries following injury. TBI: Traumatic brain injury.
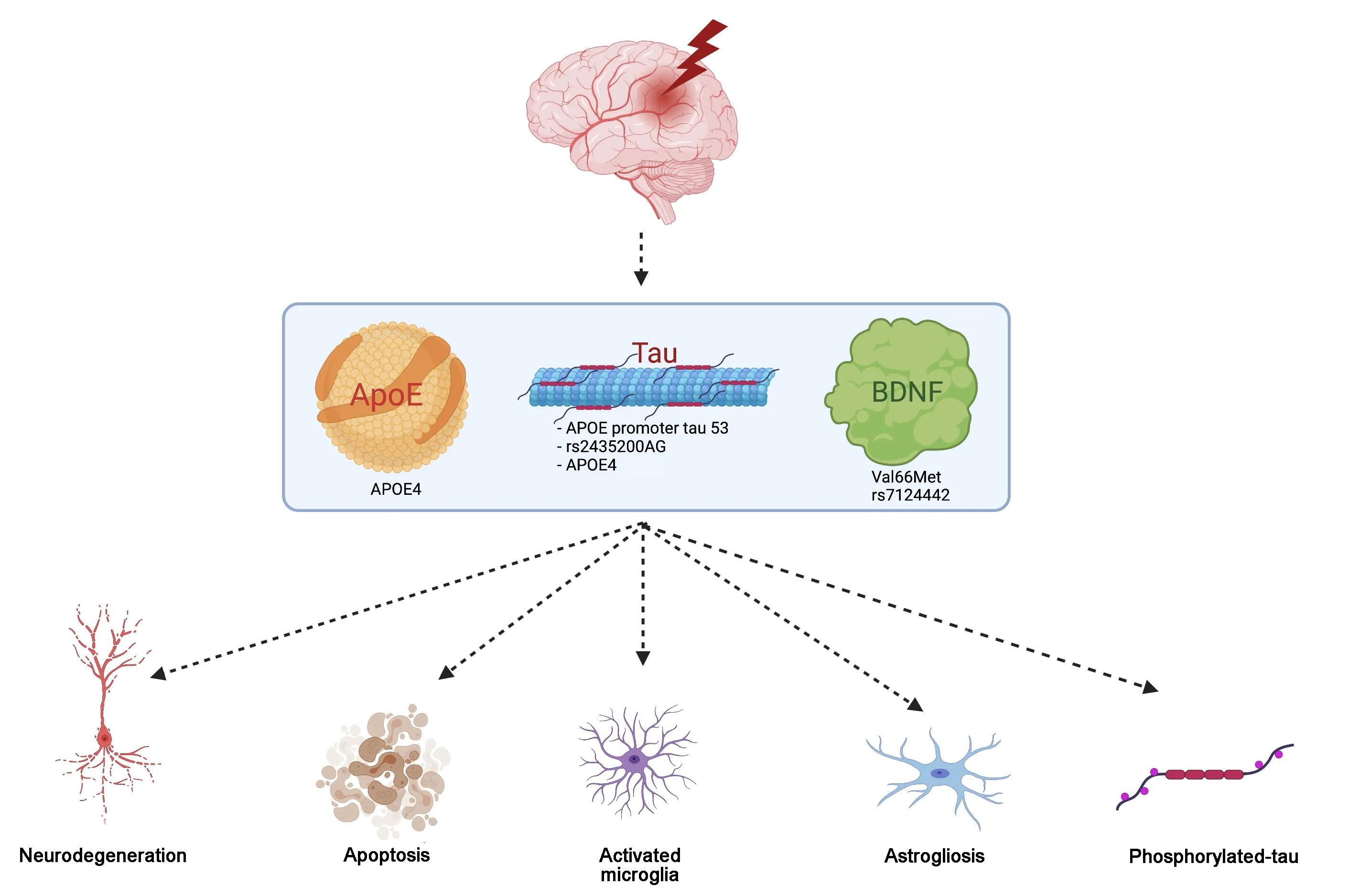
Figure 2 | Genetic risk factors and their impact on TBI outcomes.Individuals with the certain genetic risk factors are more susceptible to poor outcomes following TBI including increased neurodegeneration, apoptosis, activated microglia,astrogliosis and phosphorylated tau. These genetic risk factors involve polymorphisms within the APOE, Tau and BDNF genes - APOE4, APOE promoter tau 53, rs2435200AG,Val66Met and rs7124442. APOE: Apolipoprotein E; BDNF: brain-derived neurotrophic factor; TBI: traumatic brain injury.
Brain-derived neurotropic factor
In addition to the neurotrophin BDNF serving as a potential biomarker for TBI,there may be genetic polymorphisms in the BDNF gene that predict outcomes after TBI. BDNF has numerous important functions in the adult brain,including inducing neuronal plasticity/regeneration, regulating synapses, and playing an essential role in hippocampal long-term potentiation (Lu et al.,2014). BDNF has two major forms; the pro BDNF form which binds to the p75 receptor and activates cell death signaling cascades, and the mature BDNF form that results from cleavage of the pro BDNF form, which binds to the TrkB receptor and activates cell survival pathways as well as long-term potentiation associated with learning and memory (Alder et al., 2016). Due to the important roles that BDNF plays in brain functioning, deficits in BDNF signaling have been shown to contribute to the pathogenesis of numerous psychiatric and neurological diseases.
A common SNP in the BDNF protein occurs at the 66 amino acid position, in the pro domain of the protein. The canonical valine (Val) is replaced with a methionine (Met), in a SNP that is often referred to as either rs6265 or the BDNF Val66Met SNP. The prevalence of the Met allele ranges from 0-72%across world populations (Petryshen et al., 2010). This SNP in humans has been correlated with deficits in functions known to be modulated by BDNF activity, such as hippocampal-dependent memory as well as hippocampal plasticity (Egan et al., 2003). In anin vitromodel, neurons transfected with the Val66Met SNP showed impaired sorting of the BDNF protein as well as decreased activity-dependent release (Egan et al., 2003). Given that this SNP seems to result in less BDNF in dendrites, and less BDNF released into the synapse, it logically follows that carriers of this SNP will be at higher risk for a number of psychiatric and neurological diseases where BDNF function is important, such as diseases with impaired memory like Alzheimer’s disease(Lim et al., 2016). numerous psychiatric and neurological diseases.
In view of the role that BDNF plays in neuronal survival and plasticity, it seems likely that the Val66Met SNP may affect neuronal recovery after TBI.However, current evidence is not quite clear on whether Val66Met represents the risk allele in BDNF or if, counterintuitively, the Val66Val SNP does. Animal studies offer a way to investigate the effect of genetic polymorphisms in a controlled environment where genetic background and injury type can be precise. Our lab has used a repeated mild TBI paradigm in mice utilizing a lateral fluid percussion model that has been shown to mimic clinical TBI in human populations in a replicable manner in order to study the effect that the BDNF Val66Met SNP has on recovery. Using both male and female mice at a young middle-aged time point, we demonstrated that Val66Met carriers had increased area of inflammation, increased cell death, neurodegeneration,p-tau, astrogliosis, and activated microglia at 1 and/or 21 DPI in the cortex and hippocampus relative to Val66Val carriers. We also showed impaired learning and memory as assayed by the Morris Water Maze paradigm. Interestingly,we found that the Val66Met SNP affected both the total levels of BDNF found in the cortex at 21 DPI as well as the ratio of pro/mature BDNF found in the hippocampus at both 1 and 21 DPI with Met carriers having less total BDNF and a higher ratio of pro to mature BDNF (Giarratana et al., 2019).
When investigating this question in the human population, studies have shown that neurocognitive performance is impaired in humans with the Val66Met SNP after mild TBI relative to Val66Val carriers (Narayanan et al.,2016), as well as the fact that having the Val66Met SNP seems to be a risk factor for sustaining a mild TBI for soldiers (Dretsch et al., 2016). However,some studies have shown that contrary to previous hypotheses, Val66Val carriers actually had worse recovery than Val66Met carriers did. In a study that looked at long-term outcomes in male Vietnam combat veterans who had sustained frontal lobe penetrating TBIs, Val66Met carriers on average had significantly higher scores in a number of general cognitive functioning tests relative to Val66Val carriers (Barbey et al., 2014). Other studies have seen no effect of the Val66Met SNP on recovery from a severe TBI as measured by recovery from a vegetative state (Bagnato et al., 2012).
To complicate the matter, the Val66Met SNP is not the only SNP of clinical interest within the BDNF gene. Another BDNF SNP, rs7124442, has been associated with cognitive recovery after TBI where there is a T replacement of a C affecting neural BDNF mRNA trafficking. One group conducted a prospective longitudinal cohort study in patients with severe closed head injury TBI. They created a BDNF genetic risk score (BDNF-GRS) using the two SNPs of interest to determine how these SNPs interacted with other factors to predict outcomes after injury. While they predicted that the Val66Met and C-allele rs7124442 alleles would be the high risk BDNF-GRS, in the acute stage they saw the opposite; that the predicted no risk allele group actually had the lowest survival probability (Failla et al., 2015).
Given the contradictory results from these studies investigating BDNF SNPs as risk factors following TBI, future studies will need to explore the underlying factors at play in order to develop a cohesive theory about the role of BDNF and its isoforms after injury and how BDNF interacts with other factors such as sex and age.
Precision Medicine Approaches to Traumatic Brain Injury Treatment and Diagnosis
In light of the fact that genetic factors seem to play an important role in the differential outcomes that are seen after TBI, it is logical to assume that effective future treatments will utilize the knowledge gleaned from genetic studies to develop targeted precision medicine approaches to treating TBI.To date, there have been limited clinical studies studying precision medicine techniques for TBI (Figure 3) and none has moved into clinical practice.However, there are many preclinical studies in progress that target common TBI genetic risk factors detailed in the previous section.
Apolipoprotein E
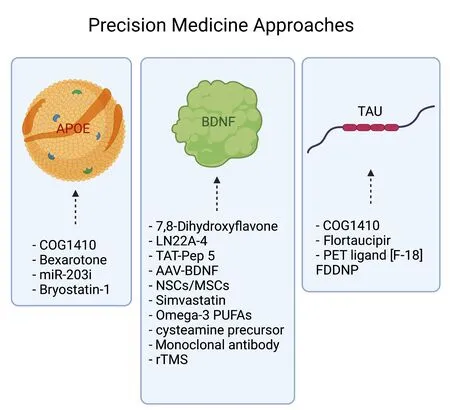
Figure 3 | Precision medicine approaches.A summary of various personalized treatments targeting APOE, BDNF and Tau genetic polymorphisms, that have been studied and are currently in clinical trial phases. AAV:Adeno-associated viruses; APOE: apolipoprotein E; BDNF: brain-derived neurotrophic factor; MSCs: mesenchymal stem cells; NSCs: neural stem cells; rTMS: repetitive transcranial magnetic stimulation.
As described above, the ApoE4 genotype is a well-known genetic risk factor associated with poorer outcomes following various types of TBI. Common targets of personalized treatments are known biomarkers which have been shown to be altered in TBI-induced animal models. An ApoE protein mimetic,COG1410, has shown promise in multiple animal studies as an effective anti-inflammatory drug therapy following TBI. One study demonstrated that COG1410 improved ApoE4-induced vestibulomotor deficits and increased cerebral glucose uptake (Qin et al., 2017), while another illustrated that it reduces acute vasogenic edema in a CCI model of TBI (Cao et al., 2016).Recently, it was shown that Bexarotone inhibits inflammatory response and neuronal apoptosis, improving neurological function of mice after TBI. Bexarotone is a member of the retinoic acid family that selectively activates retinoid C receptors which promote APOE gene transcription. In a CCI mouse model, Bexarotone significantly improved motor function and spatial memory at 20 DPI (Li et al., 2017). Furthermore, it is known that tau hyperphosphorylation and a significant increase in microRNA-203 (miR-203)are found in peripheral blood of TBI mice. Thus, one lab treated mice with an miR-203 inhibitor, which suppressed TBI induced ApoE4 expression and tau hyperphosphorylation, rescuing hippocampal long-term potentiation deficits as well as hippocampus-dependent learning and memory dysfunction (Zhao et al., 2021).
Another approach to ApoE-based personalized treatments is to target signaling pathways that have a differential response in the E3 genotype compared to the E4 genotype. One such signaling pathway is protein kinase C epsilon(PKCe). Apolipoprotein E3 but not E4 prevents loss of synaptic networks produced by amyloid B oligomers. Thus, it was hypothesized that specific PKCe activators such as 8-(2-(2-pentyl-cyclopropylmethyl)-cyclopropyl)-octanoic acid methyl ester and Bryostatin-1, protect against this synaptic loss,whereas PKCe inhibitors block this synaptic protection by apoe3 (Sen et al.,2012). A novel personalized treatment approach in our lab treated APOE4 susceptible transgenic mice by administering Bryostatin-1, highlighting the importance of gene-specific therapies. After multiple 20 μg/kg intraperitoneal injections of Bryostatin-1, it was shown to reduce excessive inflammation and neurodegeneration in injured APOE4 mice back to the levels present in injured APOE3 mice at 1 DPI. In addition, Bryostatin-1 improved fine motor balance at 7 DPI, learning at 2 DPI, and memory at 7 DPI back to levels seen in APOE3 injured mice, demonstrating its clinical utility as a TBI therapeutic drug(Giarratana et al., 2020).
Current ApoE and TBI-based clinical trials examine mild TBI and Alzheimer’s disease in patients via cognitive function and amyloid positron emission tomography (A-PET) study. One novel study (NCT01871610) linked mild TBI and Alzheimer’s disease by A-PET and cognitive function tests at the 1-yeartime point. The study aims to determine the importance of APOE genotypes of amyloid accumulation and cognitive impairment and later shed light on further clinical studies and amyloid clearing therapy for prevention and treatment for dementia after mild TBI. Another currently recruiting clinical trial (NCT02134041) hopes to study the effects of mild TBI on accumulation of amyloid beta via A-PET. A-PET could be a significantly helpful tool for understanding real impacts and pathophysiological mechanisms of mild TBI on Alzheimer’s disease.
Tau
While there has not been a substantial amount of research done investigating the role that tau genetic polymorphisms play after TBI, there has been a significant amount of research into the role that tau, especially phosphorylated tau, plays in increasing neurodegenerative sequelae after TBI.In relation to this, it has also been shown that increases in phosphorylated tau have been found in APOE4 carriers (Li et al., 2017; Giarratana et al., 2020).Therefore, one targeted treatment approach may be to attempt to reduce levels of tau after TBI, especially in carriers of high-risk genotypes, such as APOE4 carriers.
Animal models have used an ApoE-based therapeutic, COG1410, in APOE4 carriers in order to ameliorate the neurodegenerative pathology seen after TBI. In addition to improving ApoE4-induced vestibulomotor deficits (Qin et al., 2017) and reducing edema (Cao et al., 2016), studies have been done to show that COG1410 treatment decreases levels of phosphorylated tau(Laskowitz et al., 2010).
Clinical trials have mainly focused on tau as a neuroimaging biomarker to determine the risk stratification of people who have sustained a TBI. There have been 44 clinical trials registered involving the investigation of tau in TBI. Of the 11 of these that have been completed, 4 have posted results. The first of the completed studies reporting results was done in retired National Football League (NFL) players who had clinical symptoms that were consistent with a diagnosis of CTE (the diagnosis of which can only currently be made postmortem) in an attempt to find a PET tracer that could assist in making the CTE diagnosisin vivo. Because tau accumulation is used to make the CTE diagnosis postmortem, the research team used a PET tau ligand, flortaucipir,in their study. They found that former NFL players had significantly higher standardized uptake value ratios for the tau ligand than the control group of former non-contact athletes in three brain regions (Stern et al., 2019).Another completed study used the tau PET ligand [F-18] FDDNP to investigate the same question, and consistent with other neuropathological theories of injury, they found that tau pathology is seen in subcortical, limbic, and cortical areas in a different pattern from the neurodegenerative changes that are seen in Alzheimer’s disease (Barrio et al., 2015). Notably, they were able to compare the findings from one of their subject’sin vivoimaging to the postmortem brain analysis, to confirm that the [F-18] FDDNP-PET binding levels were correlated with tau deposition in the postmortem brain,suggesting that this ligand may be effective at diagnosing CTEin vivo(Omalu et al., 2018). These results highlight the use of tau as a potential biomarker that can be used for both diagnostic purposes, as well as a target for personalized treatment approaches in higher risk individuals, such as APOE4 carriers, who may have an elevated tau burden after TBI.
Brain-derived neurotrophic factor
As discussed above, BDNF plays an important role in synaptic plasticity and regeneration and it has become evident that genetic polymorphisms can cause differential outcomes after TBI, potentially due to differences in BDNF expression. It has been shown that carriers of the 66Met allele have altered levels of total BDNF after injury, as well as a shift in the pro/mature BDNF ratio(Giarratana et al., 2019). As such, precision medicine approaches that aim to increase total or mature BDNF after injury may hold promise.
In order to do this, preclinical trials have focused on more direct methods to target BDNF and activate mature BDNF/trkB signaling pathways. BDNF itself is ill suited for systemic therapeutic treatment due to its short halflife and inability to cross the BBB. Therefore, pre-clinical researchers have used a number of different treatment modalities such as traditional small molecules treatment approaches, stem cell treatment options, both naked and encapsulated/on platforms, as well as viral vector delivery systems to overcome the limitations of BDNF administration.
One method uses BDNF mimics that act as trkB agonists in order to stimulate mature BDNF signaling (Massa et al., 2010). The most well studied trkB agonists in a TBI model are 7,8-dihydroxyflavone (Alder et al., 2016) and LM22A-4 (Fletcher et al., 2021), both of which have shown improvements in recovery post TBI after treatment in mouse models. An alternative method targeting BDNF signaling is to block the pro-BDNF/p75 pro-apoptotic cellsignaling pathway, in order to preferentially stimulate signaling along the mature BDNF/trkB signaling pathway. This has been done using small molecules, such as TAT-Pep5, which has been shown to reduce neural death and degeneration in a mouse model of TBI (Alder et al., 2016). Another method that is being employed to overcome the inherent difficulties of using BDNF treatment is to embed BDNF in injectable scaffolds, so that levels of BDNF remain high at the site of injury for an extended period of time. In one study, researchers used a collagen-binding domain fused with BDNF as a treatment method after TBI in a mouse model. They found that injecting this treatment into the brain alleviated the inflammatory response and improved behavioral outcomes of mice after injury (Yin et al., 2020).
Another approach to targeting BDNF signaling is to employ adeno-associated viruses (AAV) as a vector to carry the wild type BDNF gene. Using this gene therapy method, the 66Val form of the BDNF gene can be delivered to the area of injury, and wildtype BDNF will be producedin vivowithout being held back by the limitations of delivering the BDNF peptide itself. This offers a targeted solution to the problem posed by the BDNF Val66Met variant being a risk factor for poor outcomes after TBI. Our lab has shown that treatment of Val66Met mice after repeated mild TBI with AAV-BDNF increases levels of mature BDNF and phosphorylation of the mature BDNF receptor trkB, and is able to improve cellular, motor, and cognitive behavioral outcomes at 21 DPI(Giarratana et al., 2019). This is a promising new treatment method that may offer the option for precision medicine techniques for those at highest risk for poor outcomes following TBI.
In addition to these methods, biomedical engineering techniques have begun to employ the use of stem cells, such as neural stem cells and mesenchymal stem cells as treatment for TBI. Stem cells are able to act as a therapy in two primary ways, cell replacement and exosome secretion. Through cell replacement, they are able to replace and repair areas of injury. Through their secreted exosome, they are able to express beneficial factors such as growth factors and cytokines. Stem cells can either be injected to the site of injury naked, where cell replacement is their primary mechanism of action, or protected by either encapsulation or being embedded into a scaffold, where their exosome is their primary mechanism of action. For example, one group studied the effect that intravenous administration of mesenchymal stem cells had on outcomes and BDNF levels after injury. They found that mesenchymal stem cells migrated to the area of injury, where they increased levels of BDNF and Nerve Growth Factor and had improved functional outcomes. The authors hypothesized that the increases in BDNF and Nerve Growth Factor were the mechanism through which the functional improvements were made(Mahmood et al., 2004). Another group co-cultured human mesenchymal stem cells with activated astrocytes and seeded these cells into functional BDNF peptide hydrogel, which they injected into the area of injury and found that it was effective at stimulating tissue regeneration (Shi et al., 2016).
While these pre-clinical studies are promising, current clinical studies have utilized more broad-based interventions that may rely on BDNF as a mechanism for improved outcomes after TBI. For example, it has been well established that exercise results in a robust increase in BDNF in humans (Walsh and Tschakovsky, 2018), with some groups showing that aerobic exercise increases the ratio of mature-BDNF/proBDNF in the hippocampus relative to sedentary controls in a rodent model (Luo et al., 2019). Because of the wellknown effects of exercise in increasing BDNF levels, many groups have been interested in developing exercise-based interventions for treatment after TBI in order to utilize this method to increase mature BDNF levels and stimulate synaptic plasticity and repair after injury. In addition to simply exercise-based interventions, others have considered the use of a multi-model approach,adding in meditation and cognitive training to the intervention regimen.While previous studies in healthy humans have shown that aerobic exercise significantly increases serum BDNF levels, they have not shown that cognitive training nor meditation are able to increase serum BDNF levels (Chou et al.,2018).
Currently, there are five clinical trials on record that use exercise as an intervention after TBI, either alone or as part of a multi-model approach,evaluating levels of BDNF after the intervention, three of which have been completed, and however the results are still pending from these studies(NCT00619463, NCT02276079, and NCT03674398). On the other hand,preclinical research investigating the effect of exercise on outcomes after TBI showed that exercise improved cognitive function in a rat model, and that these beneficial effects were modulated by BDNF and trkB signaling (Chou et al., 2018).
In addition to the studies that use exercise as an intervention, other studies have used drug interventions and measured peripheral BDNF as a biomarker when evaluating outcomes. There are currently five trials on record using drugs as an intervention after TBI. Drugs such as simvastatin (NCT01952288),omega-3 polyunsaturated fatty acids (NCT02990091, NCT03345550), a cysteamine precursor that shows anti-inflammatory activity (NCT04262895),and a monoclonal antibody to the cis isomer of tau phosphorylated at threonine 231 (NCT04677829) are being investigated as a treatment option after TBI. Of these studies, only the study investigating simvastatin has been completed, and the study results are pending. However, preclinical research that investigated simvastatin treatment in a rat model has shown that it is able to upregulate BDNF and its signaling pathways, and results in an increase in neurogenesis and improved functional outcomes (Wu et al., 2008). There is also evidence from the preclinical literature that omega-3 polyunsaturated fatty acids increase levels of BDNF, reduced oxidative damage, and counteracted learning disability in a rat model of injury (Wu et al., 2004).
Interestingly, one completed study investigated the use of Repetitive Transcranial Magnetic Stimulation as an intervention after TBI. They hypothesized that BDNF was the mechanism responsible for changes in brain connectivity due to repetitive transcranial magnetic stimulation treatment.Specifically, they investigated the effect that the repetitive transcranial magnetic stimulation treatment modality has on pro and mature plasma BDNF levels across both Val66Val and Val66Met carriers. In their preliminary results, they found that repetitive transcranial magnetic stimulation treatment increases the mature BDNF/proBDNF ratio (NCT02152540) (Adamson et al.,2019) .
Discussion
In this review, we have addressed the current literature investigating the risk conferred by single gene polymorphisms. However, the development of a polygenic risk score (PRS), as used in many other diseases to determine the risk for a disease given the complex genetic background of humans,would be an important step in moving from preclinical research into clinical practice. A PRS is an estimate of an individual’s genetic vulnerability to a trait or disease, adjusted according to their genotype profile and relevant genome wide association study data (Roberts et al., 2019). Current risk prediction considerations include age, sex, family history, medical history, social history,and mental health history; however, there are still differences in outcomes between individuals with similar risk factors. There have been few studies recently exploring the clinical utility of PRS for major diseases. In a Finnish study, it was discovered that coronary artery disease PRS combined with traditional risk factors would be cost-beneficial if deployed in a targeted approach (Treble-Barna et al., 2020).
While no current PRS currently exists for TBI, one study attempted to create one for the analysis of persistent post concussive symptoms. To do this, they utilized PRS’ derived from GWAS of Alzheimer’s disease, Parkinson’s disease,schizophrenia, bipolar disorder, and major depressive disorder, along with the years of schooling, college completion, childhood intelligence, infant head circumference and adult intracranial volume. However, they found no association with most of the GWAS, except for the factor of high intracranial head circumference being correlated with positive outcomes. This study suggests that genetic predisposition to persistent post concussive syndrome following TBI overlaps with mechanisms related to early brain development and growth (Polimanti et al., 2017). The development of a PRS system for TBI and even various types of TBI, would allow physicians to more accurately identify high-risk individuals who will face worse outcomes following TBI and is an important next step in this research.
Conclusion
In this review, we have identified potential genetic risk factors that increase the risk for poor outcomes after TBI. These risk factors include theAPOE4and BDNF Val66Met genetic polymorphisms among others (Giarratana et al., 2019, 2020). Using these potential genetic risk factors, researchers have identified potential targets for personalized treatment approaches that reduce known biomarkers which correlate with worse outcomes after TBI.In preclinical experiments, drugs such as bryostatin-1 (Giarratana et al.,2020) and COG1410 (Cao et al., 2016; Li et al., 2017; Qin et al., 2017) have been shown to improve both diagnostic biomarkers and outcomes after TBI inAPOE4carriers, while drugs such as AAV-BDNF (Giarratana et al., 2019),7,8-dihydroxyflavone (7,8-DHF) (Alder et al., 2016), and LM22A-4 (Fletcher et al., 2021) have been used for BDNF Val66Met carriers and to target trkB signaling after TBI. Clinical trials to date have focused on less specific treatments, using drugs such as simvastatin, omega-3 polyunsaturated fatty acids, anti-inflammatory drugs, and a monoclonal antibody to the cis isomer of tau phosphorylated at threonine 231 however many of the trials are still pending results. Other interventions have included exercise and repetitive transcranial magnetic stimulation to increase BDNF levels, which have preliminary promising results (Su et al., 2019).
To conclude, in this review we attempted to highlight the status of preclinical and clinical research into precision medicine approaches for diagnosis,prognosis, and treatment for TBI. We stress the importance of further research into this area, in order to improve the clinical efficacy of treatments for TBI in the future.
Author contributions:Manuscript conception and design, literature search,manuscript writing: SR, AG; intellectual collaboration and assistance: JA, STV.All authors approved the final version of this manuscript.
Conflicts of interest:None declared.
Availability of data and materials:All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Nathan K Evanson, Cincinnati Children’s Hospital, USA;Stuart H Friess, Washington University, USA.
Additional file:Open peer review reports 1, 2.

- 中国神经再生研究(英文版)的其它文章
- Functional in vivo assessment of stem cell-secreted prooligodendroglial factors
- iGluR expression in the hippocampal formation, entorhinal cortex,and superior temporal gyrus in Alzheimer’s disease
- Exploiting Caenorhabditis elegans to discover human gut microbiotamediated intervention strategies in protein conformational diseases
- N-methyl-D-aspartate receptor functions altered by neuronal PTP1B activation in Alzheimer’s disease and schizophrenia models
- Aminopeptidase A and dipeptidyl peptidase 4: a pathogenic duo in Alzheimer’s disease?
- Ubiquitin homeostasis disruption,a common cause of proteostasis collapse in amyotrophic lateral sclerosis?