
Unraveling pathological mechanisms in neurological disorders: the impact of cell-based and organoid models
2022-03-09 07:33JakeLanglieRahulMittalArielFinbergNathalieBencieJeenuMittalHosseinOmidianYadollahOmidiAdrienEshraghi
中国神经再生研究(英文版) 2022年10期
Jake Langlie, Rahul Mittal, Ariel Finberg, Nathalie B. Bencie, Jeenu Mittal,Hossein Omidian, Yadollah Omidi, Adrien A. Eshraghi,3,4,5,*
Abstract Cell-based models are a promising tool in deciphering the molecular mechanisms underlying the pathogenesis of neurological disorders as well as aiding in the discovery and development of future drug therapies. The greatest challenge is creating cell-based models that encapsulate the vast phenotypic presentations as well as the underlying genotypic etiology of these conditions.In this article, we discuss the recent advancements in cell-based models for understanding the pathophysiology of neurological disorders. We reviewed studies discussing the progression of cellbased models to the advancement of three-dimensional models and organoids that provide a more accurate model of the pathophysiology of neurological disorders in vivo. The better we understand how to create more precise models of the neurological system, the sooner we will be able to create patient-specific models and large libraries of these neurological disorders. While three-dimensional models can be used to discover the linking factors to connect the varying phenotypes, such models will also help to understand the early pathophysiology of these neurological disorders and how they are affected by their environment. The three-dimensional cell models will allow us to create more specific treatments and uncover potentially preventative measures in neurological disorders such as autism spectrum disorder, Parkinson’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis.
Key Words: Alzheimer’s disease; amyotrophic lateral sclerosis; autism spectrum disorder; cell-based model; central nervous system; induced pluripotent stem cells; mesenchymal stem cells; organoid model; organ-on-a-chip; Parkinson’s disease; three-dimensional model
Introduction
and the development of novel therapeutics.
Stem cells, including embryonic stem cells (ESCs), tissue-derived mesenchymal stem cells (MSCs), and induced pluripotent stem cells (iPSCs), have become an increasingly popular area of research showing promise in new treatments and providing fundamental knowledge of pathologies. Cell-based models have been utilized to understand the safety and determine the molecular mechanisms of new drug therapies and to model diseased states (Mittal et al., 2019). These cell-based models can provide a greater understanding and in-depth appreciation for various diseases as they allow us to observe the progression of disease and real-time response to treatment. There are also challenges in acquiring biopsies from live patients. With respect to the central nervous system, it is arguably unethical and dangerous to obtain multiple biopsies from the same patient to understand the progression of disease.Therefore, cell-based models allow us to begin to understand the progression from healthy to pathological tissue in numerous diseases.
The advancement of cell-based models has not only allowed us to recapitulate the normal physiology but has also allowed us to begin to replicate components of neurological diseases. These neurological diseases include amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD),Alzheimer’s disease (AD), and autism spectrum disorder (ASD) (Eshraghi et al.,2018; Mittal et al., 2019). As we begin to understand more of the molecular underpinnings and genetic differences of these diseases, we are better able to model the pathophysiology in cell-based models using new technologies such as single-cell imaging and gene editing (Horvath et al., 2016). The cell-based models of these diseases have necessarily advanced beyond two-dimensional(2D) cell cultures and include new cell-based assays in drug discovery and pathophysiology studies. This includes iPSCs, three-dimensional (3D) cocultures of multiple cell lines, and organ-on-a-chip systems (Horvath et al.,2016). Replicating individual diseased cell lines and combining them with functional models of other cells into a 3D model best mimics the diseased state that existsin vivo. Molecular techniques have allowed us to understand genetic and epidemiological changes in addition to the regulation of proteins and transcription factors present in the disease. To best utilize cell-based models, incorporation and modeling of accessory structures, extracellular fluids, and non-sensory cells are essential. This review aims to impart on recent advances in the cell-based modeling of neurological disorders (including ASD, PD, AD, and ALS) while discussing the pathophysiology of these diseases
Database Search Strategy
Referenceswere compiled using PubMed, Cochrane Library, and Medline databases. The search included articles published up to 2021 in this narrative review. A wide array of keywords and their abbreviated versions were used,including Alzheimer’s Disease (AD), Amyotrophic Lateral Sclerosis (ALS),Parkinson’s Disease (PD), autism spectrum disorder (ASD), cell-based models,organ-on-a-chip, induced pluripotent stem cells (iPSCs), human pluripotent stem cells (hPSCs), mesenchymal stem cells (MSCs), pathophysiology, etiology,drug treatment, therapeutics, and human embryonic stem cells (hESCs). Peerreviewed articles were selected that: (1) emphasized advancement of cellbased and organoid models, and (2) highlighted a unique finding or obstacle in modeling neurological disorders.
Cell-Based Models
Mimicking neurological disorders using anin vitromodel has been the goal of researchers for many years. Such models, if designed to mimic thein vivocondition, allow us to better understand pathological mechanisms and molecular alterations as well as test the efficacy of various drugs before advancing toin vivotrials (Figure 1).In vitromodels offer the advantage of reduced animal use, higher throughput, reduced cost, and better control of the physical and chemical environment.
2D models

Figure 1 |Applications of cell-based models.Cell models for neurological systems can be derived from patient somatic cells, converted to iPSCs, and driven to various cell lines based on exogenous transcription factors and proteins. Specific neurons that could fall under neurons include dopaminergic, GABAergic inhibitory and glutaminergic stimulatory neurons. The cells can then be used for a variety of applications including cellular therapy, transplantation studies, 3D Models,genetic screening, drug exploration, organoid models, and toxicity studies. Created with BioRender.com.
Protocols have been developed to establish human embryonic-like stem cells through the reprogramming of somatic cells using stem cell technologies.The basis for this technology is rooted in a study by Yamanaka et al who used a retroviral vector to exogenously express four transcription factors,Oct3/4,Sox2,Klf4, andc-myc, that drove somatic fibroblasts back to a state of pluripotency (Takahashi and Yamanaka, 2006). Some of the first iPSC models generated were of neurogenic diseases, including ALS, PD, and AD(Dimos et al., 2008; Soldner et al., 2009; Israel et al., 2012). An alternative cell-based model of neurological disorders includes direct harvesting of ESCs from the blastocyst and differentiating to the desired cell line. However, these models are much more difficult to harvest and generate compared to iPSCs(Simmnacher et al., 2019).The differentiation of iPSCs into neurons with various neurotransmitter phenotypes provides new opportunities to model central nervous system(CNS) diseases (Tao and Zhang, 2016). With the generation of iPSC models and recent developments in the generation of organoids, we are beginning to see tissue-based modeling of many neurodegenerative disorders including ASD, PD, ALS, and AD. 2Din vitromodels characteristically allow for high replicative potential with minimal cost to the researcher when compared toin vivoor 3D organoid models. This can allow for large-scale drug or toxin screening before advancing to a more complex model. However, limitations to 2Din vitromodels include possible cross-contamination with other cell types,the limited doubling potential of cell lines, polarity, and the possibility of infection of the culture medium. In addition, 2D models do not closely mimic the natural organization of tissues.
3D models
Recent work in the field of modeling neurodegenerative diseases has focused on 3D organoid models of the brain, where they can transplant multiple cell lines of generated iPSCs into one model (Di Lullo and Kriegstein, 2017; Benito-Kwiecinski and Lancaster, 2020). There have been multiple cerebral models generated, from preliminary derivation of the pituitary and cerebral cortex A progressing to much more complex models that include all six cortical layers of the forebrain (Qian et al., 2016). Models have even recapitulated the human brain after the first trimester (Lancaster et al., 2013). There are multiple approaches to generate these brain organoids. Figure 2 highlights the overall pattern of brain organoid generation through the use of iPSCs and ESCs to generate whole-brain or specific regional organoids.
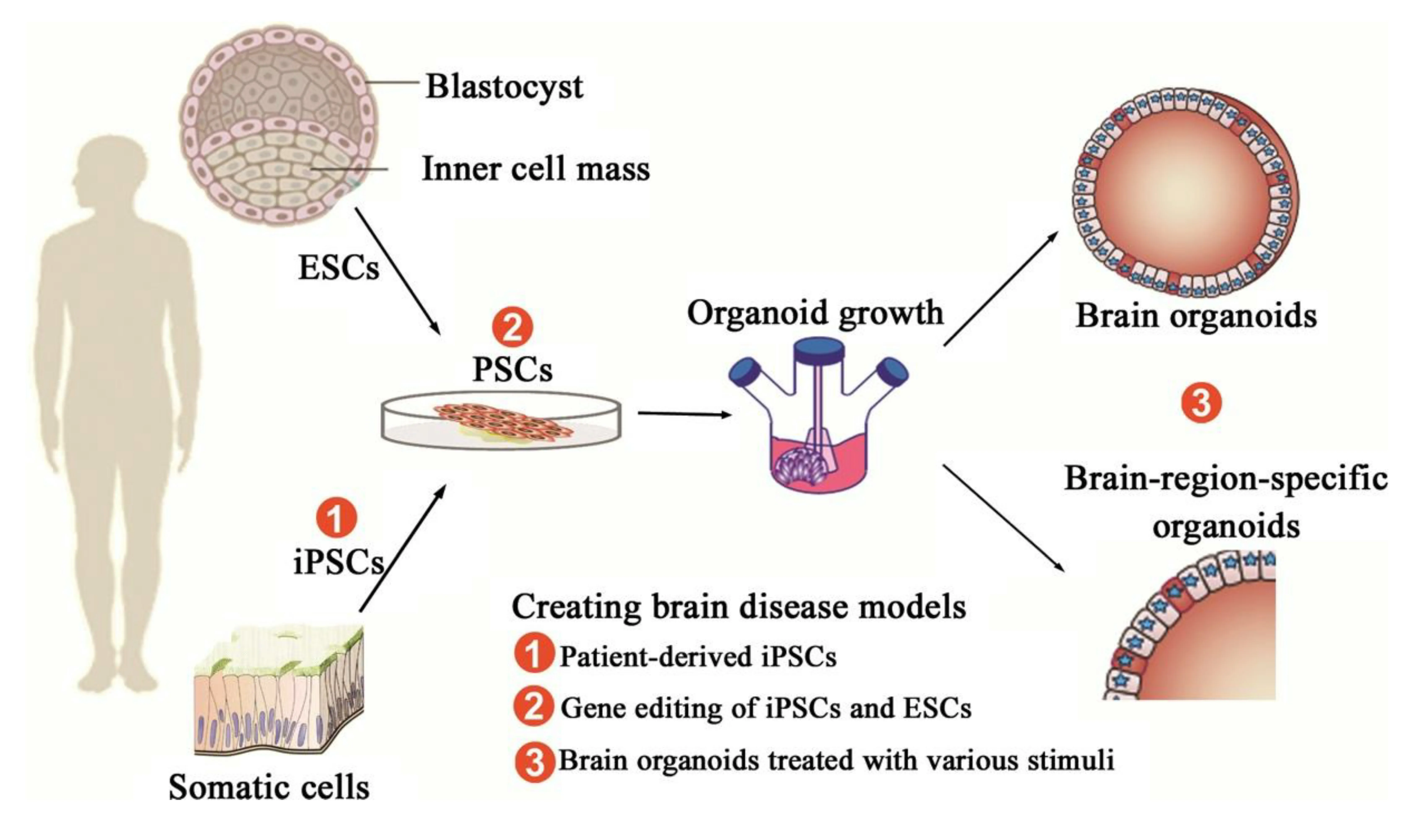
Figure 2 |Generation of brain organoids and brain disease models.Pluripotent stem cells (PSCs) include embryonic stem cells (ESCs) and induced PSCs(iPSCs). ESCs can be generated from a blastocyst, and iPSCs can be reprogrammed from somatic cells. Brain organoids are induced and differentiated from PSCs successively through different developmental stages, and eventually maintained in the spinning bioreactor. PSCs can develop into brain organoids by virtue of self-organization and differentiate into brain-region-specific organoids with the use of inducing factors. As for diseased brain organoids, three kinds of methods are used to create certain brain disease models: (1) using patient-derived iPSCs that are reprogrammed from patient-derived somatic cells, such as skin cells and blood cells; (2) applying gene editing to modify disease-linked alleles of PSCs, including ESCs and iPSC; (3) treating brain organoids with various stimuli, such as oxygen-glucose deprivation for an in vitro ischemic stroke model.Taken from an article originally published in CNS Neuroscience and Therapeutics in 2017 by Wang et al. (2017). Reprinted with permission from Wiley Online Library.
Matrigel matrix model of 3D organoid generation
A majority of models utilize serum-free culture to generate embryoid body-like aggregates. These are induced with forebrain inductive signals,growth factors, and extracellular matrix proteins to induce growth of the brain (Mariani et al., 2012; Kadoshima et al., 2013; Ilieva et al., 2018). The generated neurospheres are subsequently embedded in Matrigel that is terminally differentiated within a spinning bioreactor.
To begin, iPSCs are cultured for roughly 5 days until they become embryoid bodies through germ layer differentiation. The embryoid bodies are then further differentiated into neurospheres through neural induction. These neurospheres are subsequently embedded within a Matrigel hydrogel droplet. Spontaneously due to the embedding process, budding neurospheres generate fluid-filled cavities that are similar toin vivoventricles. The neuroepithelium then beings to migrate outwards, generating cortical layers consisting of layered neural progenitor cells. With subsequent exposure to retinoic acid, cerebral organoids are able to self-organize through selfpatterning mechanisms to populations of neural progenitors including radial glia, expanding the cerebral structures. As the progenitor cells are grown within a spinning bioreactor, radial glia differentiate and migrate to form the cortical plate consisting of neurons, the subventricular zone consisting of basal radial glia and intermediate progenitors, and the ventricular zone,named for its location adjacent to the ventricle, composed of apical radial glia. The neuronal migration depends on a layer of Cajal-Retzius cells and the outward migration is regulated by the protein reelin secreted by the latter cells (Frotscher, 1998). This technique to generate CNS cerebral organoids is highlighted in Figure 3.
Neuronal layering method of brain organoid development
Pluripotent stem cells (PSCs) include embryonic stem cells (ESCs) and induced PSCs (iPSCs). ESCs can be generated from the blastocyst, and iPSCs can be reprogrammed from somatic cells. These cell lines can then be transplanted into a matrix to allow for distinct neuronal layering in culture. This can allow the researcher to mimic the layers of cell lines that are present in normal physiology. Protocols have been developed for the generation of cortical identity with neuralization occurring spontaneously. However, exogenous factors including WNT inhibitors, preventing the differential into non-neural ectoderm and mesoderm, and are often used in combination with BMP inhibitors of TFG-beta inhibitors to maintain a neural identity (Chiaradia and Lancaster, 2020). Generation of brain organoids often has overlapping approaches, but the general goal is to successfully mimic thein vivophysiology of the normal cortical structures.
Advantages of 3D models
Cerebral organoids allow us to advance beyond ourin vitrocell culture models to more closely mimic normal physiology. To limit the amount of animal use,these structures can give us an intermediate bridge betweenin vivomodels and 2Din vitrocultures. 3D models allow for a higher sense of complexity when compared to their 2D counterparts. There is a need for expansion of 3D organoid models as they do not require the extensive maintenance and cost ofin vivomodels, can be scaled to a high level, and allow for greater control of the model to tease out underlying mechanisms. The 3D model can then be advanced to anin-vivomodel.
Cell-Based Models of Common Neurological Disorders
In this section, we discuss cell-based models of common neurological diseases and highlight the new technology of organoid-based and 3D models in the field of neurodegenerative diseases. We will conclude by highlighting the limitations, challenges, and solutions of 3D organoid models.
Autism spectrum disorders: cell-based models Background of autism spectrum disorders
ASD comprises an array of neurodevelopmental deficits that affect how a person communicates and interacts with others. This spectrum includes the previously characterized diagnoses of Asperger’s disorder, pervasive developmental disorder, childhood disintegrative disorder, and autistic disorder (Hodges et al., 2020). The key drivers of pathogenesis indicate early changes in neurogenesis and network formation within the nervous system(Acab and Muotri, 2015). While transgenic and knockout mouse models have been created, they typically target only one gene. With a heterogeneous population of possible candidate genes and a known pleomorphic disorder such as autism, it is not feasible to model the disease in its entirety in an animal model (Acab and Muotri, 2015). The advancement of iPSCs that allow the generation of a library of stem cell models from any individual has paved the way to replicate disease and patient-specific disorders. These models can help to understand the molecular and cellular mechanisms driving ASD.
2D in vitro models of autism spectrum disorders
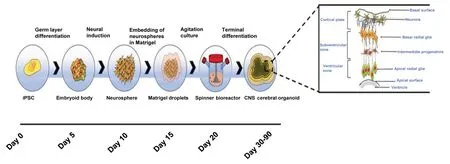
Figure 3 |Cerebral organoid formation.One way to generate three-dimensional organoid models of the brain is using iPSCs that are differentiated into neurospheres and subsequently embedded in Matrigel. These neurospheres are then growth and terminally differentiated within a spinning bioreactor, generating radial glia that is patterned through the use of retinoic acid and proteins secreted from Cajal-Retzius cells to generate CNS cerebral organoids. Taken from Pacitti et al. (2019), https://www.frontiersin.org/articles/10.3389/fncel.2019.00129/full. This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited (Pacitti et al., 2019). CNS: Central nervous system; iPSCs: induced pluripotent stem cells.
The hope for the development of iPSCs in the field of autism is to generate patient-specific iPSCs to replace nonfunctional neurons in the CNS (Acab and Muotri, 2015). However, in generating iPSCs, it is difficult to understand which phenotypes represent the diseased state as there is a deficit of knowledge about the underlying mechanisms driving neurodegeneration in ASD. One study showed that beyond genetic mutations, ASD is modulated by some environmental and external factors such as cytokines that mediate neurogenesis in the womb, specifically heat shock proteins (Lin et al., 2014).There are known gene mutations generated in iPSCs that are correlated with ASD includingSH3, neurexin 1 (NRXN1), multiple ankyrin repeat domains 2/3 (SHANK2/3), and neuroligin 3/4X (NLGN3/4X) (Gouder et al., 2019). The underlying alteration of synaptic homeostasis coded by these genes could represent a uniting process that can correlate them with ASD (Prilutsky et al.,2014). It has also been known that some genetic syndromes, such as Dup15q,cause phenotypes such as motor deficits, hypotonia, moderate to severe intellectual disability, and speech and language development. These deficits often qualify the individual for a diagnosis of ASD. These mutations can cause hyperexcitability of neurons and offer new electrophysiological targets for the treatment of known genetic causes of ASD (Fink et al., 2018).
ASD has no clear etiology, and the genetic heterogeneity provides a limited understanding of the pathways. However, a large number of the genes that have been linked to autism share common functions including neurotransmitter pathways (Trobiani et al., 2018; DiCarlo et al., 2020), neuron adhesion, dendrite size, and spinogenesis (Deshpande et al., 2017; Gouder et al., 2019), calcium-regulated signaling (Schmunk et al., 2017), neuronal hyperexcitability (Fink et al., 2018; Deneault et al., 2019), and junction molecules (Fiorentino et al., 2016; Lutz et al., 2020).
Many of the models generated have focused on syndromic autism. However,nonsyndromic autism, known as classic autism, has also been replicated in an iPSC model. Autism typically presents with poor social responsiveness and delayed development of motor skills (Griesi-Oliveira et al., 2020). Recent models have attempted to mimic nonsyndromic autism, but few have shown success.
Given that autism comprised a spectrum of disorders, two alternative 2D models have been proposed to model the disease. The first model is to use a single patient who demonstrates a characteristic clinical manifestation of autism, and subsequently monitor the differences in the molecular and cellular signature compared to a healthy and matched control. Although this pathway does not fully encompass the heterogeneity of autism, if repeated,it would allow for identification of a group of target genes and pathways.An alternative solution is to use a larger subset of patients presenting with idiopathic ASD and generate organoids from these patients, allowing us to distinguish pathological findings from controls in a 3D model (Ilieva et al.,2018). Rather than creating a representative iPSC model of autism, it may be more useful to first generate a library of ASD iPSCs with the capability to tease out the molecular pathways affected by different individuals and identify specific phenotypes. This could be followed by advancing to a larger scale organoid model. The key cell-based models of autism spectrum disorder and related disorders are reviewed in Table 1, highlighting the mutation, cell line,and objective of the model.
3D organoid models of autism spectrum disorders
Due to the heterogeneous nature of the etiology of autism spectrum disorder,at the current time and knowledge of the disease, it is difficult to create an organoid model that accurately captures the features of the disease. The tradeoff between clinical manifestations and molecular underpinnings needs to be balanced to generate a representative brain organoid model of ASD (Benito-Kwiecinski and Lancaster, 2020).
However, organoid models have been generated of diseases known to be associated with autism including neonatal infection of Zika virus, leading to microcephaly (Vianna et al., 2018; Wheeler, 2018; Santi et al., 2021). Thus,brain organoid models have been generated to model exposure to Zika virus and its effects on neurodevelopment (Qian et al., 2016). Zika often causes microcephaly, and therefore, brain organoid models mimicking microcephaly may help us to deduce neurological deficits that appear during development,leading to cognitive abnormalities that also appear in individuals with ASD(Lancaster et al., 2013).Brain-specific organoids can also be generated of disorders that induce syndromic forms of ASD with known genetic causes. These include Angelman syndrome, which has a high comorbidity with autism and shares a common genetic basis with some forms of ASD (Khatri and Man, 2019; Sun et al.,2019). Key 3D models of ASD are reviewed in Table 2.
Amyotrophic lateral sclerosis: cell-based models
Background of autism spectrum disorders
As a paralytic and neurodegenerative disorder, ALS is characterized by the sequential degeneration of motor neurons in the CNS (Brown and Al-Chalabi,2017). There are no treatments to stop or reverse the effects of the disease.Therefore, most patients have a mean survival of three to five years following initial diagnosis (van Es et al., 2017). Around 10% of diagnosed cases are due to familial-inherited forms of ALS, while the remaining 90% of diagnoses are from sporadic cases. Various molecular pathways underlining the genetic heterogeneity of the disease have been implicated, encompassing around twenty genes. These genes code for cellular signaling defects, surrounding non-neuronal cell dysfunction, and abnormal protein aggregation (Van Damme et al., 2017). SOD-1 mutant familial ALS is the most common subset of genetic mutations in ALS patients, but specific treatments have yet to be fruitful (Fujimori et al., 2018). Rodent models have been generated of ALS, yet these models are not easily translatable due to the complexity of the human CNS. One study showed that the lifespan of ALS mice could be improved by transplanting neural progenitor cells derived from human iPSCs, a possible method of regeneration to be exploited in future human clinical trials (Kondo et al., 2014).
2D models of amyotrophic lateral sclerosis
Other than organoid-based models, iPSC models of ALS appear to be ideal for mimicking human-specific neuronal and glial cell dysfunction. As multiple etiologies of ALS have been discovered, there have also been multiple cellbased models of ALS generated in recent years. In one study, iPSCs were created from patients with ALS and subsequently differentiated into glial cell types and motor neurons specifically affected in ALS (Dimos et al., 2008).These cells could have future uses in elucidating the mechanisms of disease and serving as screening models for novel therapeutics. A more recent study modeled a common subtype of ALS, ALS8, using patients that presented with a vesicle associated membrane protein associated protein B (VAPB)mutation (Mitne-Neto et al., 2011). When compared to controls, mutant ALS8 neurons displayed reduced protein levels of VAPB in an iPSC model, thereby implicating a mechanism of defective transport in the pathogenesis of ALS.As the number of iPSC-ALS models expands, we expect new drug discoveries in the field, including the identification of novel therapeutic agents based on libraries of culturedin vitroALS models (Fujimori et al., 2018). The key cellbased models of ALS are reviewed in Table 3, highlighting the pathogenetic and mechanistic findings discovered through the models.
3D models of amyotrophic lateral sclerosis
It appears that advancement from an iPSC model to an organoid model that can recapitulate ALS on a tissue-level is not far from reality. A recent study successfully generated an organoid of motor neurons from stem cells, thereby generating a model of the principal cell affected in amyotrophic lateral sclerosis patients (Kawada et al., 2017). Functional human neuromuscular junctions of sensorimotor neurons in organoid models have also been developed from gene-edited neurons created to harbor familial ALS mutations as well as derived from native ALS iPSC lines (Pereira et al., 2021). These models mimic the impairment present at the neuromuscular junction in ALS.There has also been a successful generation of microglia that innately develop in a cerebral organoid model, adding to the complexity of the model needed to appropriately represent ALSin vivo(Ormel et al., 2018). As we continue to expand cerebral organoids and increase their complexity, we may potentially reach a representative model of ALS needed to elucidate its etiology and develop a targeted therapy (Vieira de Sa et al., 2021). The key 3D models of ALS and related motor neuron disorders are reviewed in Table 4.

Table 1 |Neurological Cell-based iPSC models for Autism spectrum disorder

Table 2 |Neurological 3D and organoid models for autism spectrum disorder and related diseases

Table 3 |Neurological cell-based iPSC models for amyotrophic lateral sclerosis

Table 4 |Neurological 3D and organoid models for amyotrophic lateral sclerosis and related motor neuron dysfunction
Parkinson’s disease: cell-based models
Background of Parkinson’s diseasePD is a neurodegenerative disorder of movement that displays the pathognomonic death of dopaminergic neurons of the substantia nigra.There is an increased incidence of PD in association with aging, as it is most commonly observed in elderly populations. Clinical presentations of PD include rigor, bradykinesia, and tremor. Two classifications of PD exist, with 90% of cases occurring sporadically and only around 10% of cases due to familial causes of PD. To date, most of the research in the field has focused on familial classifications of common pathological variants of PD, includingSNCA,LRRK2,PINK1, andPRNK2(Simmnacher et al., 2019). The etiology for sporadic PD is largely unknown, limiting its modeling, but environmental toxins (e.g., heavy metals and pesticides) are believed to be important in the pathogenesis of the disease (Ball et al., 2019). The current models of PD focus on the generation of dopaminergic neurons derived from ESCs, neural stem cells, or human iPSCs, allowing for the mimicking of pathological processes and molecular changesin vitro(Zhang et al., 2017).
2D in vitro models of Parkinson’s disease
Modeling PD through a stem cell-based approach is difficult, given that multiple phenotypes of the disease exist, and each requires a separate cell line to be modeled. However, creating a library of iPSCs of PD to model both sporadic and familial phenotypes may be key to (i) elucidating novel molecular pathways, and (ii) understanding the regulation that plays into the Parkinson’s phenotype. PD is a unique CNS disorder, whose characteristics do not arise until the late stages of the disease. This adds to the complexity of modeling the disease as aging dopaminergic cell lines are difficult to model. Early iPSC modeling of PD was shown to successfully create dopaminergic neurons,yet the key cellular phenotypes indicative of PD failed to emerge (Soldner et al., 2009). Those changes may not have emerged due to inadequate aging of the model, or because the changes were too subtle to be detected by methods current at that time. However, various entities have been used to mimic the cell stress experienced by PD cells, including inducible factors like growth factor deficiencies, mitochondrial toxins (Ryan et al., 2013), or even accelerating aging through the modulation of progerin, a protein known to be involved in premature aging (Miller et al., 2013).
Most models have focused on familial cases of PD includingSNCA(Byers et al., 2011; Devine et al., 2011; Mazzulli et al., 2011; Chung et al., 2013) andLRRK2(Nguyen et al., 2011; Sánchez-Danés et al., 2012; Orenstein et al.,2013; Reinhardt et al., 2013). These models have shown that dopaminergic neurons in PD are adversely affected and susceptible to oxidative stress withLRRK2mutations (Jiang et al., 2012). The most common mutation in PD is the mutation ofp.G2019S-LRRK2in dopaminergic neurons (Nguyen et al., 2011).Mutations in alpha-synuclein (SNCA), the primary component of the hallmark features of Lewy dendrites and Lewy bodies, have also been mimicked inin vitromodels (Xu et al., 2015). Triplication ofSNCAis known to cause an aggressive and fully penetrant form of PD with dementia (Devine et al., 2011).To gain a better understanding of the interplay between the abnormal and normal structures in patients with PD, it is essential to move beyond a 2D model and into an organoid-based system. The key cell-based models of Parkinson’s disease are reviewed in Table 5, highlighting novel cell-based models.
3D models of Parkinson’s disease
The human midbrain is an essential structure to replicate in PD organoids,as many of the hallmark characteristics of the disease including Lewy bodies and Lewy dendrites arise there (Schwamborn, 2018; Kwak et al., 2020;Nickels et al., 2020). New approaches have streamlined the methods of generation of midbrain organoid generation while mitigating batch variability(Nickels et al., 2020). Standardized approaches have successfully replicated the human midbrain and have been used to study toxin-induced PD (Kim et al., 2019; Smits et al., 2019; Smits and Schwamborn, 2020). However,there is a continual need to expand the models to mimic both the genetic and environmental dispositions that facilitate the development of PD(Schwamborn, 2018). Researchers have successfully modeled idiopathic PD,through the generation of iPSCs, by transduction with Sendai viral vectors,and have directed differentiation into an organoid-like structure. They found that the expression of multiple neuron markers varied when comparing the healthy organoids with those of the PD organoid. Notably, there was a significant difference in beta-III-tubulin positive neurons, and those neurons from the PD organoid did not express tyrosine hydroxylase (TH) (Chlebanowska et al., 2020).
SpontaneousLRRK2mutations have been generated in 3D organoid systems,giving new possibilities of generatingin vivophysiology and architecture to be used for disease modeling and drug screening in PD (Kim et al., 2019).We have included a representative figure from an article by Kim et al. (2019)showing the generation of brain organoids from a specificLRRK2model of PD(Figure 4).
Recently, it has been proposed that signs of pathology are evident in the gut of PD patients far before any neurological deficits are evident. Therefore,it has been suggested to study PD using a multi-organ culture system that includes different brain regions beyond the midbrain, including organoids of the intestine and gut as well as immune cells (Reiner et al., 2021). As organoids become more developed and ornate, there is hope that further elucidation of the pathology in these models will one day lead to curative treatments for PD. Key organoid and 3D models of PD are reviewed in Table 6,highlighting the structures that could be replicatedin vitro.
Alzheimer’s disease: cell-based models
Alzheimer’s disease background
AD has been notoriously difficult to diagnose early due to its high prevalence in older populations, making it difficult to distinguish between normal agerelated cognitive decline and pathological loss of memory. AD is hypothesized to be caused by altered processing of the amyloid-beta precursor that is thought to function in the formation of pathogenic plaques in the cerebrum.A definitive diagnosis requires a post-mortem examination of the brain to identify amyloid-beta and tau protein deposits that make up the plaques and tangles pathognomonic of AD, protein deposits that were first identified by a group of psychiatrists over a hundred years ago (Goedert and Spillantini,2006). However, using current techniques for clinical evaluation of the disease, physicians can diagnose AD with 95% accuracy based on clinical evaluation and history alone (Desikan et al., 2009). Approximately 95% of cases of AD are found to be sporadic, with only 5% of cases of familial AD diagnosed annually (Desikan et al., 2009). The most common early symptoms of AD are a gradual loss of episodic memory and loss of short-term memory,eventually leading to dementia and dramatic loss of cognitive function (Mucke,2009).
2D in vitro models of Alzheimer’s disease
As elusive as the disease remains, it has been essential to create cell-based models, including iPSCs, to understand the currently unknown etiology of AD. The present limitations in understanding the underlying etiology have been difficulties in differentiating neurons already in their permanently differentiated state in sporadic forms of AD, as well as obtaining live neurons from patients (Israel et al., 2012). In a transgenic mouse model of AD, neural stem cells with upregulated BDNF were implanted and showed marked improvement in cognition (Blurton-Jones et al., 2009). However, this study has yet to yield translatable protocols to human models.
Familial forms of the patient-derived iPSCs, with increased phosphorylated tau and amyloid-beta 42:40 ratio, have been created in several models (Yagi et al., 2011; Shi et al., 2012; Kondo et al., 2013; Mahairaki et al., 2014; Penney et al., 2020). These models have been used for drug screening including β-secretase inhibitors and γ-secretase inhibitors (Yahata et al., 2011), have elucidated the increased sensitivity of AD neurons to cell death (Duan et al.,2014), have implicated mutations in APPV717I as causative for amyloid-beta accumulation and tauopathies in AD (Muratore et al., 2014) and shown that PSEN1 (Sproul et al., 2014) can play a role in the pathogenesis of AD. PSEN1 encodes the catalytic subunit of the gamma-secretase that cleaves the amyloid precursor protein. Without functional gamma-secretase, APP protein can accumulate within neurons due to improper degradation. A study successfully mimicked the elevated amounts of amyloid-beta peptide and created the pathogenic deposition of Aβ peptidein vitro(Israel et al., 2012). Models have since advanced to mimic the disruption of synaptic plasticity present in AD through extracellular forms of tau and amyloid-beta proteins (Hu et al., 2018).Models have even been able to mimic protective mutations in APP against the development of familial Alzheimer’s disease (Maloney et al., 2014). The key cell-based models of AD are reviewed in Table 7.
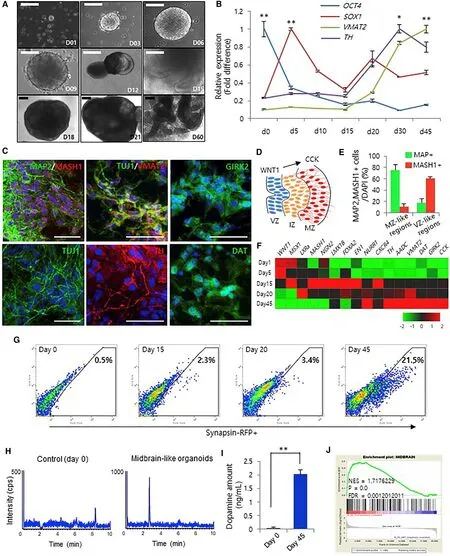
Figure 4 |Generation of midbrain 3D organoids from hiPSCs.(A) Bright-field microscopy images of the stages of 3D organoid generation for 2 months (scale bars: 200 μm).(B) qRT-PCR analysis of a pluripotency marker (OCT4),a neural progenitor marker (SOX1), and dopaminergic neuronal markers (TH and VMAT2) at different time points. Data represent the mean ± SEM (*P < 0.05, **P <0.01). (C) Immunofluorescence for MAP2, MASH1, TUJ1,VMAT2, TH, DAT, and GIRK2 to confirm the presence of midbrain dopaminergic neurons on day 60 (scale bars:50 μm). (D) Schematic image of midbrain development.The marginal zone (MZ) contains midbrain dopaminergic neurons that differentiate from radial glia cells in the ventricular zone (VZ). (E) Percentage of MAP2/MASH1-positive cells in the MZ- and VZ-like zones on day 60. (F)Gene expression profiling using qRT-PCR from 1 to 45 days. Red and green represent higher and lower gene expression levels, respectively; n = 3 per sample. (G)Fluorescence-activated cell sorting analysis of synapsin-RFP-positive cells from midbrain 3D organoids. (H and I)KCL-induced dopamine levels in midbrain 3D organoids using liquid chromatography-mass spectrometry analysis.Data represent the mean ± SEM (**P < 0.01). (J) Gene set enrichment analysis of the microarray data from midbrain 3D organoids compared with that of 2D cultures. Reprinted from Kim et al. (2019) with permission from Elsevier.
3D models of Alzheimer’s disease
Recent organoid models and organ-on-a-chip models have emerged of AD,allowing greater elucidation of the etiology, physiological representation for drug screening, and increased mimicking of the actual disease. Two forms of 3D cultures have been created in the modeling of AD, including neuronal cultures and organoid cultures. Choi et al. (2014) generated 3D differentiated familial AD neuronal cultures showing that beta and gamma secretaseinhibitors inhibit amyloid-beta and tau protein pathology. Gonzalez et al.(2018) recently generated a model of AD cerebral organoids from familial AD patients as well as iPSCs derived from individuals with Down Syndrome patient-derived iPSCs. This model showed the pathologic accumulation of amyloid-beta peptides and created structures that closely resembled amyloid plaques (Gonzalez et al., 2018).
Now, models are attempting to use multi-tissue organoid-based models including astrocytes, oligodendrocytes, microglia, and neurons. These models more closely mimic the pathology of AD, recapitulating axonal cleavage resulting from neurotoxic activities, microglial recruitment, tau hyperphosphorylation, neuroinflammatory activity, release of nitrous oxide with deleterious effects on AD astrocytes and neurons, and β-amyloid aggregation (Park et al., 2018; Papaspyropoulos et al., 2020). This model and other key 3D and organoid models of AD are reviewed in Table 8.Challenges, limitations, and solutions in generating neurologic and central nervous system organoids
To recapitulate neurological disorders, it is essential to generate reproducible protocols that can generate brain organoids with minimal variability. It has been shown that the current neurological models suffer from batch syndrome leading to significant variability in the regions as well as the quality of the models produced (Renner et al., 2017; Ilieva et al., 2018).
Another limitation is that current brain organoid models have difficulty progressing and maturing in their differentiation (Lancaster et al., 2013).In vivo, there is a time-dependent differentiation of the brain, with the primary generation of deeper layers and the subsequent development of the upper cortical layers later during the brain’s development (Harris et al., 2015).Models that rely on the self-organization of the organ have led to variable results in terms of size and structures produced in the organoid. However,some of this has been overcome through differentiating stem cellsin vitroand generating neurons in layers to mimic the different differentiation points that create a mature brain (Gaspard et al., 2008; Shi et al., 2012).
Some iPSC models require feeder cells that have a limited lifespan in culture.These cells also increase the risk of the introduction of infection to the cell culture media and lead to difficulty in mimicking the precise culture conditions between batches (Lancaster et al., 2013; Yoon et al., 2019). However, many iPSC lines now can be grown in Matrigel, providing sufficient signaling molecules to allow for successful culture without the need for feeder cells.Matrigel is a sterile substance and therefore, significantly reduces the risk of infection and allows for greater homogeneity between batches.
Organoid models of the brain that rely on self-organization have recently combined new techniques and biomaterials such as orbital shakers, spinning bioreactors, and microfluidics to overcome barriers of random neural patterning as well as allow for more homogenous models with sufficient perfusion (Kadoshima et al., 2013; Lancaster et al., 2013; Qian et al., 2016).
To progress current organoid models of the brain, limitations that do not allow for complete vascularization of the organoid need to be addressed.Insufficient maturation of neurons leads to insufficient oxygen and nutrient supply due to vascularization limits (Lancaster et al., 2013; Di Lullo and Kriegstein, 2017). Incomplete vascularization does not allow for the complete generation of the blood-brain barrier, limiting the applicability of drug treatment studies in organ-on-chip models (Huch et al., 2015).
However, incomplete vascularization has recently been addressed in both 3D models and organ-on-a-chip with successful and robust vascularization. One of the first techniques to overcome this challenge came through the ectopic expression of human ETS variant 2 (ETV2) through genetically engineered hESCs, contributing to a complex vascular-like network in human corticalorganoids (Cakir et al., 2019). Other researchers have utilized co-culture of human umbilical vein endothelial cells with hESCs or hiPSCs to create extensive vascularization within cerebral organoids (Shi et al., 2020). Recent studies have also attempted to utilize advances in bioprinting to incorporate cells of interest, including the endothelial cells of blood vessels, into largescale matrices (Grebenyuk and Ranga, 2019). Innovative techniques have also emerged of an initial culture of nonvascularized brain organoids and transplantation of the nonvascularized organoids into a Matrigel matrix laden with endothelial cells derived from iPSCs. After weeks of culture, vascularized whole-brain organoids emerged in culture (Pham et al., 2018).
These models displayed neoangiogenesis and many characteristics of a blood-brain barrier. These vascularized organoids matured more quickly and theoretically provide a much more accurate model of normal physiology.
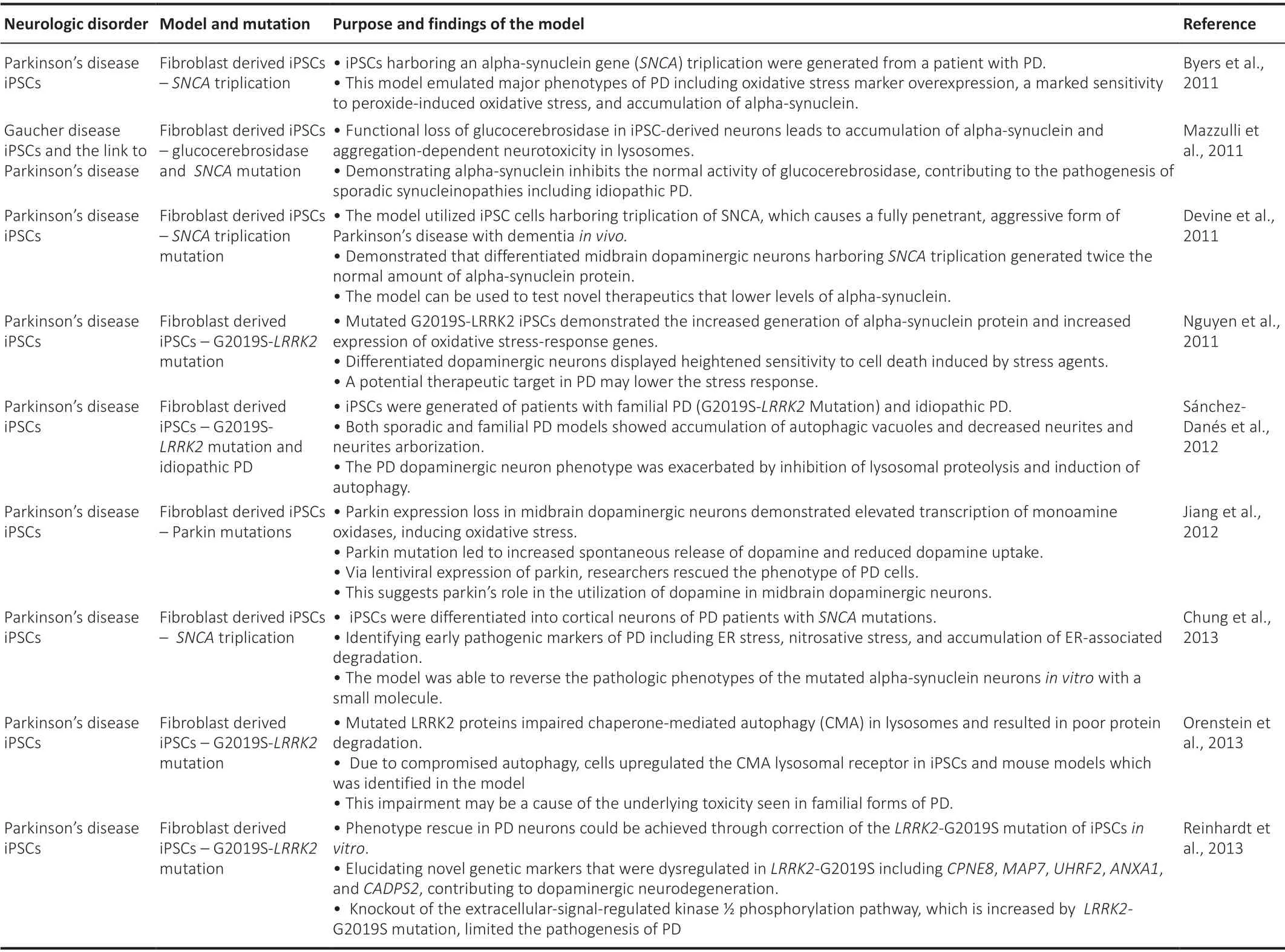
Table 5 |Neurological cell-based iPSC models for Parkinson’s disease

Table 6 |Neurological 3D models for Parkinson’s disease and related neurodegenerative models
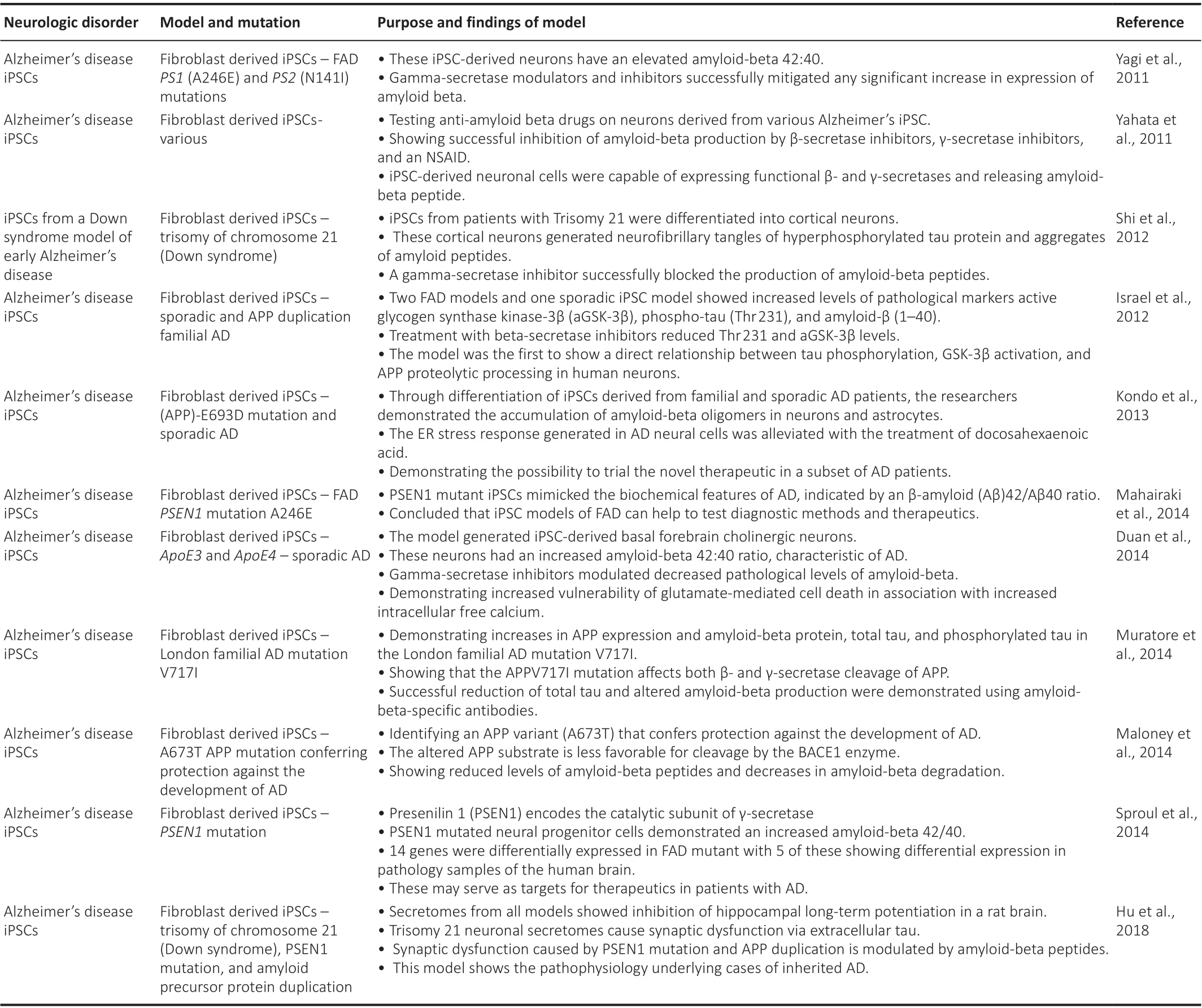
Table 7 |A summary of neurological cell-based iPSC models for AD

Table 8 |Neurological 3D models for Alzheimer’s disease
In the aging-related diseases (e.g., AD and PD) that rely on proper modeling, it is difficult to mimic the multiple genetic alterations that occur in the transcriptional profile over time (López-Otín et al., 2013). In iPSC models, we try to drive cells back to an undifferentiated state, making it rather difficult to express markers of maturity and expression while simultaneously driving cells back in their lineage (Gerakis and Hetz, 2019). This makes it difficult to mimic age-related iPSC models.
Another complication of iPSCs is that somatic cell reprogramming is extensively linked to genomic instability, especially at known tumorigenic loci (Mayshar et al., 2010). In modeling human disease in organoid models,it is essential to minimize and hopefully eliminate any undesired genetic mutations to accurately recapitulate the disease of interest.
A final limitation that has recently been overcome in organoid models is appropriate modeling and incorporation of immune cells into brain organoids(Yakoub, 2019). Without microglia, which originate directly from the yolk sac,models may not be able to fully recapitulate the normal architecture and cells of the CNS (Ginhoux and Prinz, 2015; Perdiguero et al., 2015). Emerging advancements in the field show the potential for mesodermal progenitors to develop into microglia-like cells (Ormel et al., 2018). As new models continue to emerge with microglia-containing cerebral organoids (Bodnar et al., 2021;Xu et al., 2021), we can begin to mimic thein vivophysiology more closely and hopefully, advance to better understand the pathological mechanisms of neurodegenerative disorders.
Future Directions
Through cell-based models, researchers can get a better understanding of the pathophysiology of neurological disorders.In vitromodeling can allow for the study of both normal and abnormal physiology. Organoids offer researchers another level of depth in approaching embryology and the etiology of both congenital and acquired diseases. As our understanding of these diseases begins to grow, we may begin to further stratify the pathology and develop the process of having more personalized medicine to refine our therapeutic approach to neurological disorders.
Organoid development for the neurological system is relatively new but offers hope for the regeneration of multiple cell lines that are damaged throughout the courses of these disorders. For example, in PD we are seeing the transplantation of functional and genetically corrected dopaminergic neurons that no longer display the pathological degenerative phenotype into organoidbased models. Organoid and 3D modeling offer unprecedented opportunities to provide a deeper understanding of the molecular mechanisms underlying the pathophysiology of neurological disorders.
As cell-based models begin to evolve into more organoid and 3D-based model approaches, we will eventually be able to mimic the pathophysiology of the disease, elucidate the etiologies that underpin the phenotypes of these diseases, and, hopefully, advance novel drug therapies that one day may lead to a cure for various neurological disorders.
Acknowledgments:We are thankful to Dr. Valerie Gramling from the Department of English, University of Miami, Coral Gables, FL, USA for critical reading of the manuscript.
Author contributions:All authors contributed to conceptualizing, writing,reviewing, editing and proofing the manuscript, and approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflict of interest.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Functional in vivo assessment of stem cell-secreted prooligodendroglial factors
- iGluR expression in the hippocampal formation, entorhinal cortex,and superior temporal gyrus in Alzheimer’s disease
- Exploiting Caenorhabditis elegans to discover human gut microbiotamediated intervention strategies in protein conformational diseases
- N-methyl-D-aspartate receptor functions altered by neuronal PTP1B activation in Alzheimer’s disease and schizophrenia models
- Aminopeptidase A and dipeptidyl peptidase 4: a pathogenic duo in Alzheimer’s disease?
- Ubiquitin homeostasis disruption,a common cause of proteostasis collapse in amyotrophic lateral sclerosis?