
本征缺陷对β-Ga2O3光催化性质影响的第一性原理研究
2022-03-04 13:23张静林李家印杨莲红朱应涛
原子与分子物理学报 2022年3期
张静林, 李家印, 张 龙, 杨莲红, 朱应涛
(1.昌吉学院 计算机工程系, 昌吉 831100; 2. 昌吉学院 物理系, 昌吉 831100)
1 引 言
氢气作为解决能源危机和环境污染的终极能源之一,受到科研人员的广泛关注. 其中,借助太阳能的光催化技术是制备氢气的一种重要手段. 传统半导体如TiO2,SrTiO3,因其低成本,热、化学、光稳定性高和无毒等性质,成为光催化材料研究的热点. 但是其宽带隙(大约3.0 eV),只能吸收占太阳光总能量4%的紫外光,光吸收效率低,另外光生电子空穴对具有较高的复合率,因此这些因素限制了该类半导体在光催化领域的实际应用. 为了提高光催化的效率,人们主要从两方面的工作展开:1)对传统半导体进行电子结构的调控,以提高光吸收率和光生电子空穴对分离率(比如,掺杂金属或者非金属,构建异质结等);2)寻找一种新型具有可见光吸收和电子空穴复合率低的半导体.
Ga2O3,其稳定相是β相,其他相均为亚稳相,在温度高时都会转化成β相[1]. 空间群是C2/m,其带隙在4.2-4.9 eV,本征激发只能吸收紫外光,并且是深紫外,因此该材料的研究主要集中在紫外探测器、气体传感器等. Ga2O3作为光催化材料也引起了人们极大的兴趣,但因为其宽的带隙,理想的Ga2O3光催化材料只对UV-C光有响应. 而Binet等人[2]报道了β-Ga2O3荧光谱中出现蓝光的峰,并将此归因于本征缺陷(O空位),这也为拓展光吸收的光催化研究提供了可能. 由于Ga的d10的电子组态的特点[3, 4],导带中应该是Ga原子的s或者p轨道,因此应该具有很好的光催化活性分解水产生H2和O2,Takashi等人[5]报道了Ga2O3和NiO负载的Ga2O3在深紫外区域分解水,并且解释NiO的负载是有必要的. 除了光催化分解水之外,有毒的有机废物处理也受到广泛的关注,Hou等人[6]发现多孔β-Ga2O3在紫外光区光催化分解苯的活性优于商业化的TiO2P25,可在空气气氛下将苯及其衍生物分解成为CO2,并且具有较好的稳定性. 随后他们又研究了α、β、γ相Ga2O3具有分解有机易挥发有机物的能力,得出β相Ga2O3的活性优于其他两相,并且从实验上解释了不同相光催化活性的差异源于不同的几何和电子结构. 由于β相Ga2O3导带中电子高的还原性质,Zhao等人[7]发现其可以有效降解全氟辛酸并且氟离子同时进入溶液中. 另外,β相Ga2O3可以UV-C选择性分解甲苯成为CO2,这是TiO2做不到的,作者将光催化活性高的原因归纳为高的比表面积、小的晶粒、结晶度高和定域的内电场等. 作为一种宽带隙的光催化剂,Ga2O3在紫外区表现出较高的光催化活性,太阳能的利用率不高,因此如何拓展Ga2O3的光吸收范围也引起了人们的关注. Ga2O3与中等带隙的In2O3固溶体[8]表现出高效光催化分解水的能力,可以归因于带隙的减小和带边位置的调节. 掺杂也是提高光催化活性的一个重要手段,Ryou等人[9]利用水热法制备了Sn-掺杂的Ga2O3纳米结构,表现出高的光催化活性,并将其解释为Sn作为在禁带中捕获点提高了电子和空穴对的分离. 对于非金属掺杂,N[10]和Si[11]的掺杂导致Ga2O3的吸收边红移,从而提高了其光催化的活性. Zhang等人[12]制备了Ga2O3纳米带,并指出在荧光光谱中有蓝光的发射峰,并将原因解释为在纳米带的制备过程中产生的Ga缺陷、O缺陷导致的电子结构的改变. 纳米结构材料出现的缺陷态和带边态可能使得光吸收谱发生红移,因此人们做了大量工作去研究纳米结构的Ga2O3(比如,纳米线、纳米带、纳米片)的相关性质[13-17]. 其中,Tien等人[18]制备了β相Ga2O3纳米带,光催化活性提高归因于缺陷出现导致的光吸收效率的改善. 理论上,人们对Ga2O3的几何结构、电子结构、力学性质、振动光谱及热力学性质做了大量的研究[19-22],对于β-Ga2O3及其本征缺陷引起光催化性质改变机理的解释较少. 因此,本文有必要通过第一性原理去计算含本征缺陷的Ga2O3的几何和电子性质,解释缺陷对β-Ga2O3光催化性质的影响,阐释光催化机理.
2 计算方法和细节
本文采用密度泛函的第一性原理方法计算了Ga2O3的相关几何结构和电子性质,所有的计算是在VASP(Vienna Ab initial Simulation Package)和CRYSTAL14程序中完成的. 鉴于传统广义梯度算法(GGA)低估禁带宽度的缺点,模拟中采用杂化泛函(比如B3LYP等)可以得到符合实验的禁带宽度. 而基于平面波的程序采用传统的GGA方法在几何优化中存在优势,可以得到接近实验值的几何结构参数,但是采用杂化泛函计算将非常的耗时,而CRYSTAL14采用高斯基组,在计算杂化泛函中的Fock部分相对较为容易,并且在考虑对称性的情况计算耗时较少,因此可以迅速的得到体系的电子结构,但是其对于缺少对称性的体系优化几何结构较为困难. 因此,该工作中采用传统GGA利用VASP软件进行含缺陷体系的(Ga空位和O空位)Ga2O3超胞的几何结构优化,在几何优化的基础上,采用杂化泛函基于CRYSTAL14软件进行电子结构等的计算. 下面将对两种方法采用的参数进行说明:
1)VASP,采用缀加平面波赝势方法,采用PBE(Perdew-Burke-Ernzerhof)交换关联函数和超软赝势. 平面波作为波函数展开的基组,平面波的数量是通过动量截断值(Ecutoff)来控制的,此Ecutoff值为500 eV. 对于几何优化,将所有原子上的力作为收敛的判据,设置为0.01 eV/Å,对于电子结构计算结束的标志是当能量的变化值ΔE≤1.0×10-5eV. 采用Monkhorst-Pack方案在布里渊区取点实现关于倒格矢空间的积分,对于β-Ga2O3块体网格取点大小为4×16×8,不包含Γ点. 对于含有Ga和O空位体系采用1×2×1的超胞(含有40个原子)来计算,结构如图1所示. 而对于空位缺陷,在平面波的计算就是去除原子Ga或O.

图1 β-Ga2O3几何结构图,蓝色大球和红色小球分别代表的是Ga原子和O原子. 图中标出了Ga的两种配位方式,与O形成四面体和八面体结构. 结构图由VESTA软件绘出.Fig. 1 Geometric structure of β-Ga2O3, the large blue and small red spheres represent Ga and O atoms respectively. Two coordinated types of Ga are shown in the figure, forming tetrahedral and octahedral structures with O. The structure diagram is drawn by VESTA software.
2)CRYSTAL14,采用杂化泛函交换关联函数(B3LPY),用全电子高斯函数线性组合单电子的晶体轨道,基组分别为:Ga_pob_DZVP_rev2[511s-31p-1d]和O_pob_DZVP_rev2[63311s-5311p-41d][23]. 实空间的库仑和交换积分计算使用五个参数控制(TOL1-TOL5),本文的参数值分别为10-8,10-8,10-8,10-8和10-16. 倒格矢空间取样对于块体采用8×8×8网格,对于1×2×1超胞采用4×4×4网格块体的晶格几何优化采用BFGS(Broyden-Fletcher-Goldfarb-Shanno)方案,收敛标准设置力的最大值和均方根分别小于4.5×10-4和3×10-4a.u.,而位移的最大值和均方根小于1.8×10-3和1.2×10-3,为CRYSTAL14几何优化的默认值,电子结构计算能量收敛参数设置为10-9a.u..
3 结果与讨论
3.1 理想的Ga2O3
首先,为了验证方法的可靠性并与含缺陷体系对比,本文计算了理想的β-Ga2O3几何结构和相应的性质,部分结果列于表1中. 理想的β-Ga2O3具有空间群C2/m,是单斜晶体. 晶格参数是a=12.42 Å,b=3.07 Å,c=5.86 Å,和β=103.74°(CRYSTAL14计算结果),与实验值[24, 25]和其他理论值[19-21]非常接近. 利用Brich-Murnaghan方程对E-V关系进行了拟合(如图2所示),得到了理想Ga2O3的体模量为171.75 GPa,与其他的理论计算相符合[22],因此其具有优良的力学性能. 在β-Ga2O3中,四配位的Ga与O形成四面体结构和六配位的Ga与O形成八面体结构,优化的Ga4c-O键长分别为1.85和1.89 Å,优化的Ga6c-O键长分别为1.96、2.04和2.10 Å. 因此,Ga和O并没有形成正四面体和正八面体结构,其中八面体结构中的键长稍长于四面体的Ga-O,可以推测八面体中Ga和O之间的相互作用略弱于四面体,这也在Mulliken布居分析中得到验证,Ga4c静电荷为1.650|e|,相比于Ga6c(1.615|e|),四配位的Ga失去更多的电子给O原子,Ga和O的离子键应该更强,还有Ga4c与O之间的重叠布居也大于Ga6c和O,说明了四面体中的Ga-O结合强于八面体. 图3给出了理想的Ga2O3的能带结构,导带底(conduction band minimum(CBM))位于Γ点,其价带顶(valence band maximum(VBM))出现在D和E之间的某个点(不过该点和Γ点的能量相差约0.02 eV),因此属于间接带隙结构,禁带宽度为4.73 eV,和其他的理论值相符合,也验证了其在深紫外区光作为探测器的可能性. 从能带图可以看出,VBM附近能带较为平坦,CBM附近的能带较为色散,表明其具有较低导带电子有效质量和较高的价带空穴的有效质量,电子和空穴质量较大的差异可能导致载流子的复合率降低,在不考虑光吸收的情况下,β-Ga2O3或许在光催化领域也有潜在的应用. 从总态密度(TDOS)和投影态密度(PDOS)(如图3所示)可以看出,价带顶附近主要由O的2p轨道组成,有少量Ga的s、p和d出现在价带比较深的位置,而CBM附近主要由Ga的s轨道组成,由于s轨道的离域性,因此在能带上表现出较为色散的特征,Ga的d轨道由于d10的特点,没有电子得失,出现远离VBM的位置,并且表现定域的特点,而鉴于变形的四面体和八面体结构,d轨道并没有出现明显t2g和eg的分裂,唯一例外是在八面体结构中有dx2-y2和dz2有混合,但是没有明显三下二下的分裂.

图2 β-Ga2O3的能量Energy(eV)和体积V(Å3)的关系.Fig.2 Relationship between energy (eV) and volume V (Å3) of β-Ga2O3

图3 β-Ga2O3能带结构(a)和总态密度(TDOS)及投影态密度(PDOS)(b),红色点线代表费米能级,并设置为能量的零点.Fig. 3 The band structure (a) and total density of states (TDOS) and projected density of states (PDOS) (b) of β- Ga2O3. The red dot line represents Fermi level which is set as the zero point of energy.
3.2 含Ga空位缺陷的Ga2O3
为了确定Ga和O空位缺陷的稳定性,本文计算了缺陷形成能Ef,计算公式如下:
Ef=Edef-Ebulk+μGa(μO)
其中Edef是含Ga或O空位的超胞和理想Ga2O3的总能,μGa,μO是Ga和O元素的化学势,其中μO在富氧的条件下是通过计算O2的总能得到,即,μO= 0.5EO2,对于贫氧的条件,即富Ga情况,uO=(EGa2O3-2EGa)/3,其中EGa2O3是理想的Ga2O3的总能量,EGa是bulk的金属Ga的总能除以原胞中Ga原子的数目,对于μGa反之亦然. 虽然四配位的Ga与O的相互作用比较强,但是由于配位数低,构造空位缺陷需要断键少,此处选择移除四配位Ga原子来模拟,计算的Ga空位缺陷形成能在富氧下是4.32 eV,而贫氧下为9.00 eV,O空位在富氧下为4.04 eV,而贫氧下为0.92 eV,可以看出无论在富氧或者贫氧的情况,O空位的缺陷形成能要低于Ga空位的缺陷形成能,缺陷形成能Ef越低缺陷体系越稳定,因此,推测O空位形成的可能性更大一些,由于O空位的形成,Ga2O3可能会表现出N型半导体的特征.
为了研究Ga空位缺陷对于Ga2O3体系的影响,本文计算了Ga空位体系几何结构和电子结构.Ga空位的出现虽然使得晶格参数有所减小,但是并没有引起很大的改变,最大的改变沿c轴,仅为0.09 Å. 对于Ga-O键长,只考虑了空位附近的原子的弛豫,键长有相应的缩小,并没有引起很大的变形. 因此,Ga空位的出现并未导致晶格参数和坐标明显的改变. 电子结构方面,本文计算了含Ga缺陷Ga2O3的Mulliken电荷、能带结构和态密度. 从Mulliken电荷布居分析看出,含Ga空位的Ga2O3中Ga原子的Mulliken电荷数目相较于理想情况增大,这个很容易理解是由于Ga原子的失去,多余的电子是由其他Ga原子提供. 重叠布居分析可以得出,某些Ga-O之间重叠布居增加,说明Ga-O之间的作用也有所增强,这可以解释某些键长的缩短. 含Ga空位的Ga2O3表现出顺磁性的特征,非成对电子表现出O 2p的特征. 图4给出了含Ga空位缺陷的Ga2O3能带结构,对于α自旋,CBM仍然在Γ点,VBM在Y点,保持了原来的直接带隙的特点. CBM附近能带比较色散,VBM附近能带比较定域,禁带宽度为5.08 eV,其较理想的体系有所蓝移,在禁带中没有出现杂质能级,而VBM相对于β自旋有所下降,这也是禁带宽度增加的原因. 对于β自旋,在禁带中出现了三条缺陷能带,并且均为空带,都未被电子占据,因此电子可能从Ga2O3的VBM跃迁到缺陷能级,电子跃迁能为1.93 eV,相对于理想体系的4.73 eV的禁带宽度,有显著的减小,因此空位缺陷的出现可能会导致材料的光吸收向可见光拓展,从而提高了光吸收效率. 另外,本文计算了含Ga的Ga2O3的总的能态密度(TDOS)和投影态密度(PDOS),如图4(b)所示. 从DOS和PDOS图上可以看出,和理想的Ga2O3一样,含Ga缺陷的Ga2O3的VBM只要是由O的2p轨道组成,而CBM是由Ga的4s轨道组成,因此α自旋能带保持了理想能带结构的色散特征. 只是α自旋的能带O 2p较β向低能区下移. 对于β自旋能带,Ga空位导致了禁带中缺陷能态的产生,并且这些缺陷能态主要是O 2p轨道组成. 因此,电子跃迁就是从O 2p到O 2p,由于宇称的相似性,跃迁几率可能不会太大,另外,O 2p之间的电子跃迁可能会导致电子空穴容易复合,不利于光催化反应的进行.

表1 计算的理想、Ga缺陷、O缺陷的Ga2O3几何结构(晶格参数、键长)和带隙,理想Ga2O3的体模量和Ga缺陷、O缺陷的Ga2O3的缺陷形成能.
3.3 含O空位缺陷的Ga2O3
为构建O空位缺陷体系,考虑断键的数目,本文选择移除低配位O. 优化的晶格参数a=12.43 Å,b=6.14 Å,c=5.83 Å,β=103.96°,和理想的Ga2O3相比基本没有改变. 为了观测O空位缺陷对局部几何结构影响,首先考虑缺陷附近的Ga-O键长的情况,Ga4c-O键长为1.94和2.18 Å,相对于理想的情况有所伸长,最大的伸长量为0.29 Å,而Ga6c-O键长在1.94-2.04Å之间,和理想的体系的键长改变不大,而其他位置基本上没明显的改变. 因此,和Ga空位一样,缺陷并没有引起几何结构明显的改变. 由于O原子的失去,会产生两个多余的电子,两个电子可能就会迁移到缺陷附近的Ga上,从Mulliken电荷分析可以看出,O空位附近的四配位和六配位Ga的净电荷分别为0.840|e|和0.138|e|,也验证了关于电子转移的猜测. 其他Ga的净电荷接近于理想Ga2O3. 为了研究电子跃迁和光吸收情况,本文计算了含O空位的Ga2O3的能带结构和态密度及投影态密度(如图5所示),从能带结构图上可以看出,因为O空位的引入,在Ga2O3出现了缺陷能级,并且导带边下移,此处将费米能级视为VBM,即VBM在E和C点之间,而CBM在Γ点,因此保持了间接带隙的特点,光跃迁的能量为2.92 eV,这也能解释荧光实验观察到蓝光峰的原因. 对于光吸收,Γ→Γ直接跃迁的带隙是4.30 eV,相对于理想的4.73 eV,有所减小,光吸收有红移. 和理想的Ga2O3一样,VBM附近的能带保持了定域的特点,而CBM附近的能带仍然离域,载流子的有效质量有较大的差异,也降低了载流子在迁移过程中的复合. 另外从DOS图(如图5所示)可以看出,CBM主要是Ga的4s和4p轨道占据,而价带主要是O的2p,少量的Ga 4s和4p轨道组成. 而缺陷能带(VBM附近的能带)主要是Ga 3d,4s,4p和O 2p轨道组成,表明有部分电荷转移到Ga的4s和4p轨道,这也验证了O空位出现多余的电子转移到了Ga离子上. 和含Ga空位的体系不同,电子跃迁是从缺陷态(d,s,p)到导带(4s,4p)在宇称上是被允许的. 因此,由于间接带隙、电子和空穴有效质量的差异和减小的跃迁能的特点,含O空位的Ga2O3光吸收的效率提高和光生载流子复合率降低,从而有可能提高其光催化的效率.

图4 含Ga空位Ga2O3的能带结构(a)和态密度及投影态密度(b),(a)左图对应α自旋,(a)右图对应β自旋. 红色点线代表费米能级,并设置为能量的零点.Fig.4 The band structure (a) and thedensity of states and projected density of states (b) of Ga2O3 with Ga vacancy, the left figure in (a) corresponds to α spin, the right figure in (a) corresponds to β spin. The red dot line represents the Fermi level which is set as the zero point of the energy.
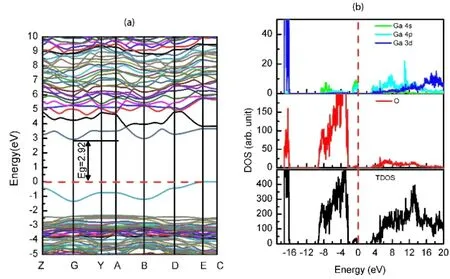
图5 含O缺陷的Ga2O3的能带结构(a)和态密度及投影态密度(b). 红色点线代表费米能级,并设置为能量的零点.Fig. 5 The band structure (a), density of states (DOS) and projected density of states (b) of Ga2O3 with O-defects. The red dots line represents Fermi level which is set as the zero point of energy.
4 结 论
基于密度泛函理论,采用标准(PBE)和杂化泛函(B3LYP),利用VASP和CRYSTAL14软件包计算了理想、含Ga空位和含O空位的几何结构和电子结构. 计算的理想的Ga2O3的晶格参数和实验相近,验证了计算的可靠性. 计算的Ga2O3的体模量为171.75 GPa,表明其具有很好的力学性质. 理想的Ga2O3属于间接带隙结构,其禁带宽度为4.73 eV,能响应紫外光,价带顶平坦的能带和导带底色散的能带表明有较大的价带空穴和较小的导带电子. 含O空位的缺陷无论在贫氧还是在富氧的条件下,形成能都小于含Ga缺陷的Ga2O3,意味着Ga2O3中空位缺陷以O空位为主,Ga2O3表现出n型半导体特点. 对于含Ga缺陷的Ga2O3,空位的出现并没有对几何结构有显著的影响,O空位的缺陷在禁带中引入了缺陷能级,电子跃迁能也降低为1.93 eV. 对于含O空位的Ga2O3体系,在空位周围的原子结构有一定的变形,键长的最大改变值为0.29 Å,晶格参数的改变很小,在禁带中出现杂质能级,电子的跃迁能为2.92 eV,相对于理想的Ga2O3有显著减小,光响应拓展到可见光区,因此Ga2O3由于其价带和导带的显著不同,在光催化材料领域有潜在的研究价值. 由于其只能吸收紫外光的特点,未来关于Ga2O3光催化的研究应该是在保持其高效的紫外光催化活性的同时拓展光吸收范围.
猜你喜欢
疯狂英语·新阅版(2021年9期)2021-10-30
小天使·聪聪画刊(2021年2期)2021-09-10
汽车零部件(2020年10期)2020-11-09
校园英语·月末(2019年11期)2019-09-10
汉语世界(The World of Chinese)(2019年6期)2019-09-10
中国科技纵横(2019年3期)2019-03-25
作文中学版(2018年1期)2018-11-28
汽车生活(2018年5期)2018-06-21
分析化学(2017年12期)2017-12-25
分析化学(2015年3期)2015-04-20
