
Diverse and precision therapies open new horizons for patients with advanced pancreatic ductal adenocarcinoma
2022-03-02 06:51RiLanBaiNanYaWangLingLingZhaoYongFeiZhangJiuWeiCui
Ri-Lan Bai, Nan-Ya Wang, Ling-Ling Zhao, Yong-Fei Zhang, Jiu-Wei Cui
Cancer Center, the First Hospital of Jilin University, Changchun 130021, China
Keywords:Tumor Pancreatic ductal adenocarcinoma Targeted therapy Immunotherapy Epigenetics Tumor microenvironment
ABSTRACT Pancreatic ductal adenocarcinoma (PDAC) is a common cause of cancer-related death, and most patients are with advanced disease when diagnosed. At present, despite a variety of treatments have been developed for PDAC, few effective treatment options are available; on the other hand, PDAC shows significant resistance to chemoradiotherapy, targeted therapy, and immunotherapy due to its heterogeneous genetic profile, molecular signaling pathways, and complex tumor immune microenvironment. Nevertheless, over the past decades, there have been many new advances in the key theory and understanding of the intrinsic mechanisms and complexity of molecular biology and molecular immunology in pancreatic cancer, based on which more and more diverse new means and reasonable combination strategies for PDAC treatment have been developed and preliminary breakthroughs have been made. With the continuous exploration, from surgical local treatment to comprehensive medical management, the research-diagnosismanagement system of pancreatic cancer is improving. This review focused on the variety of treatments for advanced PDAC, including traditional chemotherapy, targeted therapy, immunotherapy, microenvironment matrix regulation as well as the treatment targeting epigenetics, metabolism and cancer stem cells.We pointed out the current research bottlenecks and future exploration directions.
Introduction
Pancreatic ductal adenocarcinoma (PDAC), as one of the most intractable and refractory tumors, is the third most common cause of cancer-related death in the United States [1] . It is expected to become the second leading cause of cancer-related death in the United States by 2030 [1] . However, at present, less than 20% of patients with pancreatic cancer present with early disease or feasible surgical resection [2] . Most patients developed advanced or metastatic PDAC (mPDAC), a disease that is almost universally fatal,with a survival time of less than 1 year and few effective treatment options available [3]. Recently, combination chemotherapy with significant efficacy has prolonged the survival of mPDAC patients by only 2-4 months, accompanied by significant toxic and side effects, with a 5-year survival rate of less than 10% [ 4 , 5 ]. Although targeted therapy achieves significant response in some patients with specific mutations, such patients account for a very small proportion. Immunotherapy has significant efficacy in many malignancies [6-9] , but not in PDAC because most patients with pancreatic cancer have a highly complex immunosuppressive tumor microenvironment (TME), low tumor mutation burden and immunogenic antigens, and lack of high-quality effector T-cell infiltration [ 10 , 11 ],which makes the immunotherapy less effective on the treatment of pancreatic cancer. PDAC exhibits resistance to chemoradiation, targeted therapy, and immunotherapy. The mechanisms include genetic and epigenetic alterations, and complex TME. Over the past decade or so, with the rapid development of multi-omics, bioinformatics, high-throughput sequencing technology and the development of genetically engineered mice (GEM) models, there have been many new advances in the key theory and understanding of the intrinsic mechanism and complexity of molecular biology and molecular immunology of pancreatic cancer. Based on these theories, more diversified new methods of mPDAC treatment have been developed and preliminary breakthroughs have been made.The reasonable combination of therapies with different mechanisms may bring the greatest hope for mPDAC patients and have an important impact on the integrated treatment model of pancreatic cancer. Accordingly, this review focused on the progress of comprehensive treatment for mPDAC in recent years.
Conventional chemotherapy in mPDAC
The mainstay of current treatment for mPDAC is the combination of cytotoxic chemotherapy. The first-line treatment options are based on patient characteristics such as The Eastern Cooperative Oncology Group (ECOG) performance status and age. For patients who have lost the chance of surgery with a Karnofsky performance status ≥70, the two first-line standard treatment regimens are:FOLFIRINOX (fluorouracil, leucovorin, irinotecan, and oxaliplatin)and AG [gemcitabine combined with nanoparticle albumin-bound paclitaxel (nab-P)], and the former has an expected response rate and survival of 30% and 11-12 months, respectively, while the latter has 25% -30% and 8.5-11 months, respectively [ 4 , 12 ]. However,there are no large clinical trials directly comparing FOLFIRINOX with AG. Either regimen is a reasonable upfront option for mPDAC patients . The exploration for biomarkers that predict response to chemotherapy in patients with advanced disease will be an active area of future research. For example, loss of GATA 6 expression may help in the selection of the first-line regimens by distinguishing between classical and other PDAC subtypes, since classical subtypes is more likely to benefit from fluoropyrimidine-based therapy [ 13 , 14 ]. Patients with DNA damage repair (DDR) mutations may benefit from mFOLFIRINOX [15] . In addition, the FOLFIRINOX had a high incidence of hematologic, gastrointestinal, and neurotoxic side effects, while AG had an increased risk of peripheral neuropathy and myelosuppression [ 4 , 12 ]. Considering that both main regimens pose a challenge to patient tolerance, other options were explored, such as GS (gemcitabine plus S-1) regimen in Asia.Based on the current research data at the American Society of Clinical Oncology (ASCO) meeting, it is proposed that nab-P combined with S-1 regimen can be considered to replace the traditional AG regimen for mPDAC.
In the second-line setting, patients with good performance status may receive an alternative first-line regimen that has not been previously used. The only international standard secondline therapy is the combination of 5-fluorouracil/leucovorin (5-FU/LV) and nanoliposomal irinotecan (nal-IRI) for patients receiving gemcitabine-based therapy in first-line therapy. Based on the Asian results of the nal-IRI with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1) study, the Chinese Society of Clinical Oncology (CSCO) Guidelines for the Diagnosis and Treatment of Pancreatic Cancer updated this protocol to Class 1A Evidence and Level I Expert Recommendation [ 16 , 17 ]. At the latest ASCO conference, a multicenter real-world observational study in Korea compared the second-line application of FOLFIRINOX and nal-IRI plus 5-FU/LV after failure of first-line gemcitabine-based therapy [18] ,and the results showed that the two regimens have equivalent efficacy and are safe and tolerable. However, these results were not replicated in the European and North American cohorts, indicating population-specific differences. For patients with poor performance status, gemcitabine, S-1, or nab-P chemotherapy regimens are mainly considered. In addition, the recent ASCO meeting reported a study (OnFX) of bermekimab, an interleukin (IL) -1αantagonist, in combination with nal-IRI plus 5-FU/LV in patients with advanced PDAC and cachexia, and the preliminary results showed that patients in the combination group had an overall survival benefit of 10.5 months, a duration of response of more than 90%,and a progression-free survival (PFS) of 7.7 months, indicating that the new regimen was effective in cachexia patients [19] . In addition, standard combination regimen drug sequences or combinations with other drugs were explored. A quadruple regimen of AG-based cisplatin and capecitabine in 24 patients showed partial response in 16 patients and stable disease in 8 patients [20] ;one study is testing a lower dose of five chemotherapeutic agents(gemcitabine, nab-P, carbamazepine, irinotecan, and capecitabine)and is expected to have results in 2021 (NCT03535727). Therefore,it is expected to further improve the survival outcome of PDAC patients by developing novel cytotoxic agents or combination strategies, while rationally using biomarkers.
Molecular targeted therapy in mPDAC
In recent years, with the development of gene high-throughput sequencing technology, the mechanisms of genes and molecular pathways related to the development of PDAC have been gradually deepened. Integrative genomic, transcriptomic, and proteomic analysis of 150 PDAC specimens showed that somatic mutations in v-ki-ras2 kirsten rat sarcoma viral oncogene homolog(KRAS)(93%),TP53(72%),CDKN2A(30%), andSMAD4(32%) were the four most common key oncogene or oncogene mutations, but these were hardly clinically actionable genetic alterations [21] . Recently,best matched targeted therapies, such as poly (ADP-ribose) polymerase (PARP) inhibitors targeting germline breast cancer susceptibility gene (BRCA)1 orBRCA2mutations, TRK inhibitors forNTRK1,NTRK2, orNTRK3fusions, and immune checkpoint inhibitors(ICIs) for mismatch repair (MMR)-deficient or high microsatellite instability tumors, have been used in mPDAC patients. Patients with these genetic alterations account for approximately 8% of the population, but only a minority received matched targeted therapies. In the Know Your Tumor program in the United States, 189 of 677 patients were detected with actionable molecular changes, and overall survival was significantly longer in patients who received matched treatment (n= 46) than in those who received unmatched treatment alone (n= 143) [2.58 years (2.39-not reached) vs. 1.51 years (1.33-1.87); HR = 0.42, 95% CI: 0.26-0.68,P= 0.0 0 04] [22] . Therefore, it is important to perform genetic mutations testing and tailor effective treatment options based on the molecular characteristics of the tumor in each patient.
Therapy targeting KRAS mutations
KRASoncogenic mutations can be observed in more than 90% of human PDAC cases. Unfortunately, KRAS proteins are extremely difficult to target due to their high affinity for guanosine triohosphte (GTP) and/or guanosine diphosphate (GDP) [23] , and in mouse models, the resulting MAPK inhibition after KRAS inhibition (or direct blockade of downstream MEK) may further lead to activation of AKT, epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), andPDGFRαas well asAXL, resulting in ineffectiveness of such drugs [24] . Therefore, the development of clinically effective KRAS inhibitors has been challenging. The most commonKRASmutation in PDAC isKRASG12D. A novel alternative strategy to target KRAS involves the use of exosomes or small extracellular vesicles loaded with siRNA targetingKRASG12D[25] and investigated in a recent phase I trial(NCT03608631) that included mPDAC patients. TheKRASG12Cmutation is found only in 2% of PDAC, and its molecule inhibitors,ARS-1620 in preclinical models [26] and AMG510 (Sotorasib) in patients with advanced solid tumors [27] have shown initial antitumor efficacy. However, its activity in PDAC is unknown. Given the difficulty of directly targeting KRAS, therapies targeting major downstream effector pathways are developing, including the RAS-RAF-MEK-ERK and PI3K-PDPK1-AKT signaling pathways [28] .Chemotherapy combined with MEK inhibitors has been shown to synergistically inhibit tumor growth in an orthotopic mouse model of PDAC [29] , but the results in clinical trials of mPDAC patients was negative [30] . Notably, recent studies have elucidated the mechanisms by whichKRAS-mutant PDAC resists the pharmacological inhibition of ERK or MEK inhibitors on the MAPK signaling pathway and autophagy activation [ 31 , 32 ]. The authors hypothesized that simultaneous inhibition of MAPK signaling and autophagy have synergic effect in cell lines and patient-derived xenografts, and even observed regression of liver metastases and partial response in patients using trametinib (MEK inhibitor) combined with hydroxychloroquine (autophagy inhibitor) as off-label treatment [31] . The THREAD trial (NCT03825289) is currently evaluating the combination of trametinib and hydroxychloroquine in PDAC patients at different stages.
Therapy targeting DNA damage repair
Mutations in genes involved in homologous recombination repair (HRR) affect DNA double-strand break repair and lead to chromosomal aberrations. The overall mutation rate of the HRR pathway in pancreatic cancer is 10% -15% [33] . The most commonly mutated genes are ataxia telangiectasia-mutated gene (ATM),BRCA2,BRCA1, andPALB2gene (accession number NM_024675.3).ATMandATR(ATR serine/threonine kinase, the protein encoded by this gene belongs to the PI3/PI4-kinase family) are serine/threonine kinases involved in DDR signaling and their loss promotes angiogenesis and cell cycle progression [34] . In a small cohort of PDAC patients, the loss-of-function mutations ofATMpredicted shorter overall survival [35] . ATM inhibitors are currently in the early stages of clinical development in patients with solid tumors (NCT02588105 and NCT03423628), and have not shown application in PDAC patients. Cells compensate for ATM loss by upregulating ATR, indicating that ATR inhibitors may be effective onATM-deficient tumors [36]. ATR inhibitors are in early stages of development in patients with solid tumors (NCT03188965 and NCT03718091). PARP, targeting single-stranded DNA, has a crucial role in DDR [37] . Therefore, tumor cells with mutations that prevent HRR via other pathways are often exquisitely sensitive to PARP inhibitors, ultimately leading to cell death owing to a substantially reduced capacity for DDR (synthetic lethality). However,PARP inhibitors, including olaparib, talazoparib, rucaparib, veliparib(ABT-888), had different outcomes in several clinical trials for PDAC because of their different ability to capture PARP [37-40] . The phase III POLO study was designed to evaluate the efficacy of olaparib versus placebo as maintenance therapy in mPDAC patients with germlineBRCA1/2mutations who had not progressed after at least 16 weeks of first-line platinum-based chemotherapy [41] . The study met its primary endpoint PFS (7.4 vs. 3.8 months; HR = 0.53;P<0.004), with a duration of response of 24.9 months, indicating that such patients might significantly benefit from olaparib maintenance therapy. However, there was no significant difference in median overall survival (only 46% of events) between the two groups (18.9 vs. 18.1 months,P= 0.68), which may be related to the application of PARP inhibitors after disease progression in some patients in the placebo group. Advance events occurred early but were generally of low grade and most patients required no dose adjustment [41] . The study is the first successful phase III trial of a biomarker-driven treatment strategy in PDAC patients. However,the use of a placebo control may become a limitation, while in clinical practice maintenance chemotherapy may be a more appropriate control. Nonetheless, these results further highlighted the need to undergo molecular testing at the time of diagnosis for mPDAC. Notably, both the latest updated NCCN and the ASCO guidelines recommend germline mutation testing for PDAC. In addition to first-line maintenance therapy, early trials have also shown that PARP inhibitors are effective in the second-line treatment of HRR-deficient PDAC or in combination with first-line platinum therapy [42] .
The presence of germlineBRCA1/2mutations was determined in the POLO study based on the use of BRACAnalysis companion diagnostic testing, but the population accounts for only 5% -7% of PDAC [41] . Whole-exome sequencing and RNA sequencing methods have shown that germline or somaticBRCA1/2orPALB2mutations account for approximately 14% of PDAC [11] , andBRCA1/2,PALB2,ATM,CHEK2, andRAD51that encode mutations in DDRrelated protein genes amounted to 24% [11] . Small cohort studies have shown that PARP inhibitors have antitumor activity in cancer patients with somaticBRCA1/2mutations (in the refractory disease and maintenance setting) and in patients withPALB2mutation (maintenance therapy) [43] . Two phase II studies are currently evaluating the efficacy of olaparib in patients with non-BRCA DDR deficient (BRCAness) PDAC. The preliminary results from the US study showed a response of 20% (n= 11), while no response was reported from the Israeli trial (n= 21) [44].Unfortunately, the phase I study of PARP inhibitors in combination with chemotherapy, olaparib plus gemcitabine and olaparib plus irinotecan and cisplatin or irinotecan, cisplatin, and mitomycin C, in PDAC patients has been associated with toxicity concerns, including ≥grade 3 treatment-related adverse events in>80% of patients [45] .A study reported thatBRCA1/2mutant tumors had a higher frequency of high microsatellite instability (4.8% vs. 1.2%;P= 0.002),programmed death-ligand 1 (PD-L1) expression (22% vs. 11%;P<0.001), as well as a higher tumor mutation burden (8.7 vs. 6.5 mut/Mb;P<0.001) compared with patients with advanced PDAC withoutBRCA1/2mutations [46] , providing a basis for evaluating PARP inhibitors or other DDR protein inhibitors (e.g., ATR inhibitors) in combination with immune ICIs. The current phase I/II trial (NCT03404960) is evaluating the safety and efficacy of niraparib in combination with ipilimumab or nivolumab in PDAC patients. Studies in preclinical models ofRASmutant PDAC state that the mechanisms of resistance to PARP inhibition involves activation of RAS signaling pathway. Although the combination of PARP and MEK inhibition increases the degree of PARP trapping, it leads to higher levels of cytotoxicity [47] . The evidence that RAS signaling increases PD-L1 expression and downregulates type I major histocompatibility complex (MHC-I) expression [47] provides a rationale for multidrug combinations, such as the phase I trial(NCT03637491) evaluating the safety and activity of the MEK inhibitor binimetinib and anti-PD-L1 antibody avelumab in combination with the PARP inhibitor talazoparib in patients withRASmutant PDAC and other tumors. In addition, antiangiogenic agents have synergistic effects with PARP inhibitors, which lead to increased tissue hypoxia levels and down-regulation of homologydriven repair genes [48] . This combination will be further investigated in a phase II trial (NCT02498613) that includes mPDAC patients .
Therapy targeting other actionable aberrations
CDKN2Aencodesp16INK4A, an inhibitor of cyclin-dependent kinase 4/6 (CDK4/6) andp14ARF, which activatesp53. The loss ofCDKN2A(47% in PDAC) is a common mutation that increases CDK4/6 activity and decreases p53 activity for cell cycle progression [49] . The CDK4/6 inhibitor palbociclib has been shown to induce cell cycle arrest and apoptosis in PDAC [40] . Dual inhibition of CDK4/6 by palbociclib and MEK by trametinib has shown efficacy in xenograft models [50] . Palbociclib also reduced the expression ofα-smooth muscle actin in PDAC patient-derived xenograft models and can be used in combination with gemcitabine to reduce the incidence of liver metastases and prolong overall survival [40] .Two phase I clinical trials (NCT030 650 62 and NCT02501902) are evaluating CDK4/6 inhibitors in combination with PI3K/mTOR inhibitors or nab-P in PDAC patients. Other actionable mutations found inKRASwild-type PDAC patients includedBRAFmutation(2.2%),NRG1fusion (0.5%),NTRKgene fusion (0.3%), and anaplastic lymphoma kinase (ALK)amplification (0.16%) [51-53] .BRAFdeletion or insertion mutations can be found in about 1% of PDAC patients, and up to 10% ofKRASwild-type PDAC patients [54] .First-generation BRAF inhibitors (such as vemurafenib) have shown benefit in different cancer species carrying theBRAFV600Emutation [55] . In addition, BRAF dimerization may be altered by activation of the MAPK signaling pathway, which leads to increased levels of tumorigenesis [56] . Therefore, these alterations may also be responsive to drugs that target MAPK signaling pathways. TheNRG1gene fusion has been shown to be oncogenic inKRASwildtype cell lines, possibly due to activation of downstream EGFR signaling pathways. Two patients withNRG1fusion achieved partial response after EGFR-targeted therapy (afatinib or erlotinib), including 1 RET fusion (NCOA4-RET) [57] . The NRG1-bispecific antibody zenocutuzumab (MCLA-128), which binds to HER2 and HER3 receptors and blocks their interaction with the ligand NRG1 or its fusion protein [58] , was granted orphan drug status by the FDA for the treatment of pancreatic cancer in July 2020. Currently, phase I/II clinical trials (NCT02912949) are assessing the safety and effi-cacy of zenocutuzumab inNRG1gene fusion-positive tumors, including pancreatic cancer.
NTRKfusion mutations in PDAC are less than 1% [59] , and multiple TRK inhibitors are currently being evaluated in clinical trials. Larotrectinib (LOXO-101), targetingNTRKfusions, became the first small molecule approved by the FDA in an accelerated manner for the treatment of patients with solid tumors withNTRKfusion mutations, while subsequently entrectinib was also approved by the FDA for this indication. Clinically, it is the only TRK inhibitor that has been shown to have effect on both primary and brain metastatic lesions, without off-target activity. The latest ASCO-GI data reconfirmed that larotrectinib is recommended in a variety of gastrointestinal tumors (including pancreatic cancer) carryingNTRKfusion mutations [60] . The second-generation TRK inhibitor LOXO-195 has shown effectiveness in two patients withNTRKfusionpositive solid tumors whose disease has progressed on larotrectinib treatment [61] . Comprehensive molecular profiling identified 5 patients with ALK rearrangement among about 30 0 0 cases of PDAC, of whom 4 cases were treated with ALK inhibitors and 3 cases achieved radiographic response or serum CA19-9 returned to normal [62] . Currently, a phase I trial of the novel ALK inhibitor,ceritinib in combination with chemotherapy for mPDAC is ongoing (NCT02227940). Currently the main mTOR inhibitors used in clinical practice are everolimus and temsirolimus, but the clinical efficacy is not obvious in the phase I/II study of PDAC.STK11mutation may predict the response to mTOR inhibitors [63] , leading to the conduction of EVAMP study (NCT01178151) testing the activity of everolimus in patients with various solid tumors withSTK11mutations. HER2 expression was reported in up to 45% of PDAC patients, but the phase II study showed that the therapeutic activity of capecitabine combined with trastuzumab in HER2-positive mPDAC patients was limited [64] . FGFR aberrations were reported in 4% of PDAC patients [65] , but the sensitivity of FGFR-amplified patients to FGFR inhibitors is still unclear. The predictive role of other actionable aberrations in PDAC patients is not clear and may be elucidated in the future.
In addition, self-assembled peptide nanosystems can simultaneously deliver chemotherapeutic drugs and olaparib, and show strong inhibitory effects onBRCA-mutant PDAC [66] .Based on highly specific surface molecular markers of PDAC,plectin-1, researchers have constructed an efficient targeted self-assembled peptide nanocarrier co-delivery platform, peptide nanoparticles [67] , which has high drug loading, ideal kinetic characteristics, and integration of peptide targeting motifs. This platform can simultaneously target and deliver olaparib and the BET family protein inhibitor JQ1 while combining to treat non-BRCA-mutated pancreatic cancer. C-terminal binding protein-interacting protein is mainly involved in the homologous recombination repair process of DNA damage ends. Inhibition of the BET protein bromodomain-containing protein 4 (BRD4) has been found to impair resection of DNA ends, production of double-stranded DNA,and homologous recombination repair by reducing the expression of C-terminal binding protein-interacting protein [68]; Meanwhile, BRD4 inhibitors block the recruitment of proteins critical for DNA damage to damaged chromosomes, including RAD51, RPA70,RPA32, and MRE11 [68] . Therefore, it is speculated that after application in tumors, BRD4 inhibitor JQ1 can sensitize cells with homologous recombination defects to PARP inhibitors that inhibit DNA single-strand damage repair, and BRD4 inhibitor reverses the resistance of tumor cells to PARP inhibitors [69] . A study has confirmed in orthotopic and patient-derived xenograft models that targeted co-delivery of olaparib and JQ1 significantly enhances the synergistic effect of their combination therapy and induces apoptosis of more non-BRCA-mutant pancreatic cancer cells [67] . Systemic inhibition of DDR pathways is associated with severe side effects [ 38 , 70 ], but excitingly, this strategy significantly reduces offtarget effects in orthotopic pancreatic cancer and patient-derived xenograft models. Therefore, the development of nanobiotechnology opens up new ideas for cancer treatment.
Immunotherapy in mPDAC
PDAC is an immunologically "cold" tumor with a unique immunosuppressive TME, including a variety of immunosuppressive cells [e.g., cancer associated fibroblasts (CAFs), myeloid-derived suppressor cells (MDSCs), immunosuppressive tumor-associated macrophages (TGFs), regulatory T cells (Tregs)] and soluble immunosuppressive molecules [e.g., transforming growth factor-β(TGF-β), IL-10, IL-23] [ 71 , 72 ], resulting in an imbalance of immune effector cells. The TME of PDAC is predominantly myeloid-lineage,including tumor-associated macrophages (TAMs), granulocytes, and inflammatory monocytes, which is actively recruited to the TME by oncogenicKRASvia multiple chemokines during multistep pancreatic carcinogenesis [73] . These bone marrow cells are involved in the induction of immune checkpoint ligands on tumor cells to inhibit the function of effector T cell [73] , and can also promote the formation and maintenance of precancerous lesions directly by inducing EGF ligands, which in turn amplify MAPK signaling, downstream of oncogenicRAS[74] . GEM models have shown that oncogenicKRASactivates CAFs in the TME by inducing Hedgehog ligands. On the one hand, dense excessive extracellular matrix deposition by CAFs prevents T cells from entering the TME by creating a physical barrier [75] , and on the other hand, it can also indirectly promote immunosuppressive polarization of TAMs by directly secreting IL-6 or recruiting inhibitory B cells through CXCL13 [73] .Therefore, immunosuppressive TME of PDAC makes the clinical practice of immunotherapy currently available for pancreatic cancer very limited. Nevertheless, a variety of immunotherapy strategies have been designed for this unique TME, including strengthening/improving autoimmune responses (immunomodulators, monoclonal antibodies, tumor vaccines and cell therapy) and inhibiting tumor immune escape (ICIs and TME modulators), some of which have entered the clinical trial stage and preliminarily showed effects.
Therapeutic vaccines and oncolytic viruses
Therapeutic vaccines targeting tumor-associated antigens and/or tumor-specific neoantigens can induce antitumor immune responses by activating tumor-specific T cells, including whole tumor cells, peptides, proteins, and recombinant structures. One of the most widely studied vaccines is GVAX, a whole-cell vaccine composed of irradiated human allogeneic PDAC cell lines that has been engineered to release granulocyte-macrophage colony-stimulating factor at the inoculation site. Preliminary studies of GVAX showed expansion of metabombese-specific CD8+T cells [76] , leading to the development of a mesothelin-expressing Listeria monocytogenes (LM-mesothelin) vaccine for use in a prime-boost approach with GVAX, and confirmed in a randomized phase I trial that GVAX therapy combined with LM mesothelin boosting therapy was effective as both second- and third-line therapy [77] . Unfortunately, a randomized phase IIb study evaluating this combination versus chemotherapy in the second- and thirdline metastatic setting did not show an improvement in overall survival [78] . The study had shown that tumors can upregulate immune checkpoints and inhibit recruited and activated T cells after vaccination [79] , which prompted the exploration of the combination with ICIs. Phase I study of GVAX in combination with ipilimumab had shown that it induces antitumor T cell responses and prolongs patient overall survival [80] , and the study testing GVAX and the combination of GVAX and Listeria vaccination with ICIs and other combinations (including demethylating drugs and radiotherapy) is ongoing or under development [81] . In addition,dendritic cell (DC)-based vaccines can increase active molecules such as CD40 on the surface of DCs, enhance antigen presentation,as well as induce the activation, and kill pancreatic cancer cells by MHC-I "restricted" and antigen-specific CD8+T cells. DCs vaccines combined with other treatments can achieve better efficacy,such as enhancing T cell immune response and chemosensitivity when combined with gemcitabine [82] . TNF-αcombined with DCs vaccines [83] can also significantly enhance cytotoxic T cell killing of tumors. In addition, adjuvants may enhance the efficacy of therapeutic vaccination, such as altered or enhanced vaccine delivery vectors with cytokines. Small phase I/II trials have shown that adjuvanted vaccines of WT-1, mutantKRAS, and MUC1 (mucin) can generate T-cell immune responses and affect patient outcomes [84-86] . Although the results of phase IIb/III vaccine trials are negative, they contain key insights and provide a way toward therapeutic vaccines. Antigen selection is critical for the development of therapeutic vaccines, and improvements in bioinformatics techniques are rapidly achieving predictions for other high-priority antigens and neoantigens for vaccination [87] .
Oncolytic viruses can activate antitumor immunity by infecting tumor cells and inducing innate immune responses, directly killing infected tumor cells [88] , and have an indirect effect on the TME. Several different structural viruses, such as type I human herpesvirus, reovirus, and adenovirus, were used based on this approach [88] , with initial efficacy. For example, the phase II trial showed that reovirus resulted in a clinical benefit rate (reached partial response or complete response in 12 weeks) of 58% in 34 PDAC patients [89] . At the same time, these viruses have some potential to bind to other modulators of TME.
ICIs
Several clinical trials of ICIs in PDAC patients are currently ongoing, but early results have shown that ICI alone is almost ineffective in mPDAC [ 90 , 91 ], considering the unique immunosuppressive TME. In fact, MMR mutant or high microsatellite instability tumors have higher genomic instability, tumor mutation burden, tumorinfiltrating immune cells, and neoantigen production may be sensitive to ICIs [92] . Pembrolizumab was the first FDA-approved drug for MMR-deficient malignancies [93] , and therefore MMR testing is recommended for mPDAC patients [94] . In a retrospective study of 7 patients with microsatellite stable (MSS) mPDAC or cholangiocarcinoma withBRCAorRAD51gene mutation, 5 achieved clinical benefits after ICI treatment, including some patients with low tumor mutation burden, indicated that other patients involving DDR mutations may also respond to ICIs treatment. Given that the results of treatment are unsatisfactory in most PDAC patients, it may be more meaningful to combine other strategies to increase the proportion of effector T cells in PDAC TME in advance. Preclinical studies showed that IL-6 combined with anti-PD-L1 [95] or galectin-9 antibody combined with anti-programmed cell death-1 (PD-1) [96] could significantly inhibit the growth of pancreatic tumor; the tumor volume was reduced after adding anti-cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) therapy to CD40 plus chemotherapy in a mouse pancreatic cancer model, and the highest tumor regression rate was observed when adding PD-1 inhibitor continuously [97] . In addition, a variety of other immunosuppressive pathways are highly expressed in PDAC [98] , including T cell immunoglobulin-3, lymphocyte activation gene-3, and T cell immunoreceptor with Ig and ITIM domains, which were similar to PD-1, V-domain Ig suppressor of T-cell activation, a ligand similar to PD-L1 on medullary cells, which produces CD73, an immunosuppressive and pro-metastatic adenosine extracellular enzyme. Drugs targeting these targets may provide new insights. Further clinical trials to evaluate the combined activity of PD-1 antibody and/or CTLA-4 antibody or other ICIs are still ongoing, which is one of the hotspots in pancreatic cancer research at present.
Agonist immunotherapy
Antigen presenting cells (APCs) and T cell agonists are therapies under investigation that can prime and expand T cells. CD40 is a member of TNF receptor superfamily and is mostly expressed on DCs, B cells, and bone marrow cells. CD40 agonists target inhibitory bone marrow compartments, activate macrophages and polarize them to M1 phenotype; they have also been shown in preclinical studies to promote antigen presentation and efficient priming of cytotoxic T cells, but T cell activation has not been observed in spontaneous primary pancreatic tumors in mouse models [99] . Chemotherapy can improve the response of CD40 agonists by stimulating APCs with the release of tumor-associated antigens through cytotoxic death [100] , and then the combination of the two and nivolumab was found to induce T-cell-mediated tumor killing and produce immune memory and durable response in a small phase Ib study of mPDAC patients, with a response rate of 58% to first-line treatment [101] . Another phase I study of CD40 agonists in combination with gemcitabine observed enhanced long-term survival [102] , which may depend on the different dosing backgrounds of CD40. Currently, studies of CD40 agonist in combination with chemotherapy and PD-1 blockade(NCT02588443, NCT03214250) are ongoing. Other APC agonists targeting OX40, toll-like receptor (TLR) [103] , and stimulator of interferon genes (STING) [104] can activate DCs to improve T cell priming or induce direct costimulation of T cells. OX40 agonists induce direct costimulation of T cells, and their combination with checkpoint blockade has shown preclinical promise in other tumors [ 105 , 106 ], potentially leading to tumor infiltration of neoantigen-specific T cells, decreased LAG3 expression, and durable responses. While many agonistic pathways appear to be independent [107] , additive effects of STING and OX40 agonisms added to ICIs have been observed in breast cancer models [108] .
Adoptive T cell therapy
Chimeric antigen receptor (CAR)-T is a method in which T cells are collected from patients and manipulated to target specific antigens, expanded and reinfused, widely being used as an individualized therapy for genetically modified autologous T cells in patients [109] . CAR is designed for T cell activation and specific recognition of tumor antigens [110] . Several tumor antigens, such as bombesin, cluster of differentiation 24 (CD24), carcinoembryonic antigen (CEA), HER2, MUC1 and prostate stem cell antigen (PSCA)have been suggested as targets for pancreatic cancer. Mesothelin is highly expressed on tumors and can increase cell proliferation and migration [111] . In phase I clinical trials, 2 of 6 patients with chemotherapy-refractory mPDAC showed stable disease without adverse events after receiving autologous mesothelin-specific chimeric antigen receptor-T (CAR-T) cells (CARTmeso cells) [111] ,which led to the initiation of studies of CARTmeso cells in mPDAC patients (NCT03638193, NCT03323944). The trials involving CAR-T cell targeting mesothelin are ongoing in PDAC [112] . Overexpression of HER2, also known as CD340, proto-oncogeneNeu, or ERBB2,is associated with postoperative survival in PDAC [113] . Different CARs constructed, such asαHER2-743 andαHER2-4D5, have shown antitumor efficacy in mouse studies [114] . An early study is ongoing to assess the efficacy of CAR-T therapy targeting multiple factors including HER2 in pancreatic cancer (NCT03267173). CD24,a cancer stem cells (CSCs) marker, has been shown to be expressed in 72% of PDAC and to correlate with higher tumor grade [115] .Administration of CAR-T cells containing anti-CD24 single-chain Fv and CD28 extracellular domains, combined with irradiation and IL-2, abrogated tumors in an orthotopic human PDAC xenograft model [114] . The safety of CEA-specific CAR-T cells is being validated in different cancer types, including pancreatic cancer(NCT02349724). Importantly, most targets of CAR-T cells are also expressed in normal tissues and can trigger a range of side effects [ 116 , 117 ], including cytokine release syndrome, colitis, anaphylaxis, as well as neurotoxicity. For example, CEA-specific CAR-T cells can induce acute respiratory toxicity with increased IL-6 levels [118] ; HER2 CAR-T leads to targeted, non-tumor toxicity and patient death [99] . Therefore, it is necessary to select appropriate targets and vectors, such as targeting non-self-antigens or mutated self-antigens to improve the safety of engineered T cells. Preclinical studies utilizing "switchable" CAR-T cells engineered to specifically recognize tumor antigens by modifying their Fab segments can reduce their toxicity, and the activity of CAR-T cells can be controlled by dosage of a HER2-specific recombinant Fab-mediated“switch”, thus potentially overcoming the "on-target/off-tumor"problem [119] . In addition, the efficient transport of CAR-T cells to the tumor site and their limited persistence are another important clinical challenge. Lymphodepletion therapy in CAR-T cell therapy is observed to vary in level of persistence but needs to be considered for safety [120] , and the trial of bombesin CAR-T combined with CAR-T-19 is being conducted in PDAC (NCT02465983); CTLA-4 blockade at the basis of tumor-specific T cell metastasis induced T cell persistence and memory in melanoma patients [121] ; the addition of matrix-degrading heparanase or tumor-targeted cytokine receptors to CAR-T significantly improved intratumoral trafficking and antitumor response [122] .
Decreased natural killer (NK) cells number and activity have been observed in PDAC patients [123] . Therefore, reactivation of NK cells or highly active NK cell therapy may be potential therapies.Phase I/II studies use highly active NK cells to treat several tumors including PDAC (NCT03008304, NCT03634501). A clinical study is evaluating the killing activity of blood derived expanded NK cells from PDAC patients (NCT03665571); IL-15 has been shown to stimulate NK cell-mediated direct killing of human pancreatic cancer cells and stellate cells [124] , and to increase chemosensitivity. DCs also have an important role in immune surveillance, but there are significantly fewer CD141+DC1s and CD1c+cDC2s in the bone marrow of PDAC patients. Mechanistically, pancreatic cancer cells interrupt the development of interferon regulatory factor 8 dependent DCs to escape from immune surveillance [125] . A study found that the endogenous antigen-specific response of PDAC is abnormal due to the lack of cDCs [126] : neoantigen expression can lead to deterioration of the fibroinflammatory microenvironment, which in turn drives disease progression and metastasis, and facilitates expansion of tumor-promoting Th17 immune cells. However, restoration of cDCs in PDAC enhanced the activity of CD8+T cells and Th1 [126] . Therefore, the development or reintroduction of DCs may be a promising option for restoring immune surveillance systems. In aninvivomouse model, vaccination with pancreatic tumor cell-pulsed bone marrow-derived DCs induced T cell proliferation and inhibited orthotopic pancreatic tumor growth, which could synergize with gemcitabine to prolong mouse survival [127] .Activated T cells combined with gemcitabine and zoledronatepulsed DCs could stabilize disease in some patients [128] . In conclusion, restoration of tumor immunity by adoptive T cells is an important strategy.
Therapies targeting immunosuppressive cells and molecules in tumor microenvironment
Preclinical models have demonstrated that targeted receptors expressed on the cell surface [e.g., cxc chemokine receptor (CXCR)2, C -C chemokine receptors 2 (CCR2), colony-stimulating factor 1 receptor (CSF-1R)] can deplete inhibitory myeloid cells or macrophages, reduce tumor burden, improve antitumor immune effects and sensitize tumor-bearing mice to ICIs [ 129 , 130 ]. Binding of CCL2 to CCR2 (on inflammatory monocytes) promotes the generation of immunosuppressive TAMs [131] , and targeting this pathway leads to a significant reduction in the infiltration of TAMs and Tregs, and an increase in CD4+and CD8+T cells to reprogram the microenvironment. Treatment with a CCL2/CCR2 inhibitor (PF-04,136,309) in combination with FOLFILINOX enhanced chemotherapy efficacy and improved survival in mice with orthotopic tumors derived from the Ptf1-Cre promoter-regulatedKRASG12D-expressing and Trp53-deficient mice [132] and was initially effective in a phase IB trial (NCT02732938). A phase I/II study is analyzing the effect of combining nivolumab and a dual CCR2/CCR5 antagonist (BMS-813,160) with GVAX (NCT03767582). CSF-1R is a regulator of MDSCs and TAMs, and its inhibitors have been shown to eliminate TAMs, inhibit pancreatic cancer, and improve survival in several preclinical models [133] . Multiple studies assessed CSF-1R inhibitors as well as in combination with other ICIs or chemotherapy (NCT02777710, NCT02880371, NCT02760797,NCT02807844, NCT03336216, NCT03599362, NCT03697564). Inhibition of migration regulator CXCR2 of neutrophils and MDSCs in combination with CSF-1R inhibitors and ICIs synergistically improved T cell infiltration and antitumor responses [80] . Inhibition of CXCR4, a chemokine receptor that recruits MDSCs, promotes CD8+effector T cell infiltration in the TME, reduces MDSCs and further reduces circulating Tregs, while also targeting CAFs that are abundant in the extracellular matrix [134] . Combined inhibition of CXCR4 and PD-1 in mice synergistically increased tumor infiltration of activated T cells and induced apoptosis [75] . Recently published data from the phase IIA COMBAT clinical trial (KEYNOTE-202) [135] demonstrated that the novel CXCR4 inhibitor BL-8040(motixafortide) had good preliminary results when used in combination with pembrolizumab and triple-agent chemotherapy for second-line/third-line treatment of mPDAC. In the subsequent cohort 2 study of second-line treatments in 22 patients, ORR, disease control rate, and median duration of response were 32%, 77%,and 7.8 months, respectively, and were well tolerated. Other targets against macrophages include targeting leucine rich repeat, nucleotide oligomerization domain and pyrin domain-containing 3 signaling, dectin 1 and receptor-interacting serine/threonine protein kinase 1 (RIP1). Pyrin domain-containing 3 is a characteristic inflammasome sensor molecule whose deletion inhibits KRASinduced pancreatic fibrosis and improves survival in mice [136] ;RIP1 kinase is a key upstream regulator that controls the activation of inflammatory signaling and cell death pathways [137] , and a phase I/II study (NCT03681951) has been initiated to test the synergistic effect of RIP1 inhibitor (GSK3145095) with pembrolizumab.
In addition, multiple therapies target Tregs by altering TME medial signaling and chemokine-cytokine signaling. TGF-βpromotes immune evasion, epithelial-mesenchymal transition, and differentiation of T lymphocytes into Tregs [134] . Phase II study showed that gemcitabine combined with the TGF-β/Smad inhibitor galunisertib had a prolonged median overall survival compared with placebo(8.9 vs. 7.1 months), but hematologic toxicity was increased in the combination arm. The study of galunisertib plus PD-L1 antibody for mPDAC is ongoing (NCT02734160) [138] . The bispecific antibody M7824, which targets TGF-βand PD-L1, inhibits the two complementary immunosuppressive pathways through two distinct functional domains and has shown activity in phase I studies in multiple tumor types [139] . It also showed durable responses in 27%of 30 patients with biliary tract cancer, although 33% had toxicity of ≥grade 3 [140] . Finally, mogamulizumab, which can antagonize CCR4 highly expressed on the surface of Tregs, is under investigation and is expected to remove Tregs from the TME to generate a more robust antitumor immune response [55] .
Therapy targeting extracellular matrix in mPDAC
There are a large number of dense matrix components (collagen, hyaluronic acid and fibronectin), accounting for 90% of the tumor volume, produced by CAFs in the TME of PDAC, which can provide a favorable environment for pancreatic cancer cell growth,while it forms a barrier around the tumor, prevents immune effector cell infiltration and promotes tumor immune evasion. In addition, dense fibrous tissue can compress the blood vessels in pancreatic cancer tissue and limit the spread of chemotherapy,immunity and targeted drugs [141]. However, preclinical model data suggest that some components of the stroma may have an inhibitory effect on cancer cell proliferation [142] ; deletion of CAFs may promote tumors; and loss ofα-smooth muscle actinpositive cells in mouse models of PDAC leads to aggressive, undifferentiated tumors with enhanced hypoxia and reduced animal survival [143] . Although there is conflicting evidence on the role of matrix in PDAC, the combination of physical and functional matrix barrier modulators may produce better efficacy in PDAC (as detailed below), and their value in regulating the immune microenvironment remains worthy of further exploration.
Hyaluronic acid is overexpressed in the interstitium of PDAC patients [144] , and its increased content leads to reduced tissue perfusion and then vascular collapse through increased interstitial fluid pressure [145] . When chemotherapeutic drugs are combined with pegylated recombinant human hyaluronidase PH20(PEGPH20), their penetration into tumor tissues and antitumor effects can be significantly enhanced [146]. A randomized phase II trial of PEGPH20 in combination with AG for mPDAC showed that in patients with high levels of hyaluronic acid expression, the combination group degraded matrix proteins, improved intratumoral blood flow, and resulted in a significant improvement in PFS(9.2 vs. 5.2 months; HR = 0.51;P= 0.049) [147] . Similar randomized phase III HALO109-301 trial (NCT02715804) is ongoing [148] .However, disappointingly, the phase Ib/II randomized trial of a small cohort of mPDAC (SWOG1313) with the addition of PEGPH20 to mFOLFIRINOX showed a deleterious effect on overall survival(7.6 vs. 15.1 months; HR = 2.07;P<0.01) with an increased incidence of grade 3/4 toxicities including thromboembolic events and diarrhea [149] . Several phase I/II trials of PEGPH20 in combination with ICIs and other agents (NCT03634332, NCT03193190) are currently recruiting patients. The results of the randomized phase III trial of PEGPH20 may be reported shortly and may establish a new treatment mode for this disease.
Focal adhesion kinase (FAK) is hyperactivated (phosphorylated)in a variety of solid tumors, including PDAC, and is a regulator of fibrosis and immunosuppression in TME. The overexpression of FAK promotes pancreatic tumor colony formationinvitroand increase tumor growth rateinvivo[150] , while leading to Tregs and MDSCs recruitment, TAMs differentiation and CAFs activation and proliferation, which in turn inhibits the number and activity of CD8+T cell [151] . Administration of a FAK inhibitor (VS-4718) in a mouse model of PDAC improved the immunosuppressive microenvironment and survival outcomes and synergized with chemotherapy and ICIs [151] ; Similarly, the addition of a FAK inhibitor to gemcitabine plus ICIs resulted in a 2.5-fold increase in median overall survival in mice [152] . Combination therapies involving FAK inhibitors are currently under investigation, including the phase I trial of the FAK inhibitor defactinib in combination with pembrolizumab and gemcitabine (NCT02546531), the phase I/II trial of defactinib in combination with pembrolizumab (NCT02758587),and the phase II trial of the FAK inhibitor GSK2256098 in combination with trametinib (NCT02428270). In addition, it has been shown that tumors can develop resistance by upregulating theSTAT3pathway after FAK inhibition. Therefore, combined STAT3 pathway inhibition may be more successful in prolonging the antitumor response [153] .
Fibroblast activation protein (FAP) is expressed on CAFs of PDAC, which induces desmoplasia, promotes tumor growth and metastasis, and is associated with poor prognosis [154] .FAP-specific CAR-T improves survival in preclinical models of PDAC [155] ; In addition, FAP-positive CAFs are a major source of chemokines CCL2 and CXCL12 [156] , and their inhibition may simultaneously improve immunosuppressive TME. Bruton’s tyrosine kinase (BTK) is a non-receptor tyrosine kinase of the B-cell receptor signaling pathway. The BTK inhibitor, ibrutinib, can lead to reprogramming of M2 macrophages toward an antitumor M1 phenotype in a mouse model of PDAC, reduce MDSC numbers and immunosuppressive function in the TME, restore cytotoxic T cell activity, inhibit PDAC growth, and act synergistically with gemcitabine [ 157 , 158 ]. Unfortunately, the phase III RESOLVE trial, which included 434 mPDAC patients, did not show any benefit of ibuprofen plus AG in PFS or ICI [159] . Sonic hedgehog (SHH) signaling promotes the transdifferentiation of pancreatic stellate cells into myofibroblasts, and the secretion of matrix metallopeptidase and nerve growth factor [160] . A phase II trial of AG combined with an SHH signaling inhibitor (vismodegib) in 49 patients showed a response of 43% [161] . SHH inhibitors combined with gemcitabine still need further testing. Vitamin D plays a role in stromal immune regulation, and patients with vitamin D deficiency have a worse prognosis [130] . Clinical trials evaluating the effect of vitamin D agonists in combination with immunotherapy on matrix remodeling are ongoing (NCT03331562, NCT03519308). Connective tissue growth factor (CTGF) was highly expressed in preclinical models of PDAC and promoted fibroproliferative stromal reactions [162] . The CTGF inhibitor pamrevlumab in combination with gemcitabine has been shown to prolong survival in mouse models of PDAC [163] and this combination regimen was evaluated in a phase I/II trial in patients with unresectable PDAC(NCT02210559). Currently, the FDA have designated pamrevlumab as a fast-track therapy for the treatment of patients with locally advanced PDAC, and a randomized phase II trial of this drug is planned [164] . Finally, losartan, an angiotensin receptor inhibitor reduced collagen and hyaluronan production in the PDAC matrix, subsequently reducing shear stress and improving drug delivery [165] . Losartan in combination with chemotherapy, immunotherapy and radiotherapy is being evaluated in clinical trials in PDAC patients (NCT03563248, NCT04106856).
Therapies targeting epigenetics, metabolism, autophagy and cancer stem cells in mPDAC
Therapy targeting metabolism
The local metabolic microenvironment of PDAC contributes to the formation of immunosuppressive TME. First, the severe anoxic TME forces T cells to rely on glycolysis in glucose-depleted TME, leading to their dysfunction and antitumor immunosuppression [72] ; second, enhanced glycolysis leads to excessive lactate excretion by tumor cells, which creates immunosuppressive TME by inhibiting T cell and NK cell activation and promoting immunosuppressive polarization of bone marrow cells [ 166 , 167 ]; Third,amino acids (especially arginine and tryptophan [168] ) in TME play an important role in immune cell activation and memory T cell differentiation, and their availability is also a key determinant of antitumor immunity. In addition, amino acid metabolites, such as kynurenine, which is generated from tryptophan by arginine indoleamine 2,3-dioxygenase (IDO), also contribute to inhibitory TME formation [169] . On the other hand, tumor metabolic reprogramming is strongly associated with resistance to therapy,involving mechanisms that include selective activation of nonoxidative pentose phosphate pathway in PDAC and subsequent nucleotide biosynthesis pathways [170] , dysregulation of fatty acid metabolism [171] , and enhanced stemness of tumor cells [170] .Therefore, there is an increasing level of interest in novel agents that target metabolic signaling pathways in PDAC. Devimistat selectively inhibits the tricarboxylic acid cycle in tumor cell mitochondria by impairing the activities of pyruvate dehydrogenase andα-ketovalerate dehydrogenase, and can synergize with cytotoxic drugs by reducing the mitochondrial metabolic rate, resulting in reduced production of anabolic intermediates required for effective DDR. Among 20 mPDAC patients treated with devimistat plus mFOLFIRINOX, the response rate was 61% (CR 17%) and was well tolerated [172] , prompting a phase 3 study (NCT03504423). Devimistat in combination with AG is also being tested in a phase I study (NCT03435289). Cholesterol is an important component of the cell membrane, and pancreatic cancer cells, like other malignant proliferating cells, need to produce large amounts of cholesterol for their own needs. A study found that a large amount of free cholesterol in PDAC cells ensures their continuous demand for cholesterol by converting it into a storable form by sterol O-acyltransferase 1 (SOAT1) [173] . Inhibition of SOAT1 enzyme limited cholesterol storage, inhibited PDAC cell growth, and prolonged 50% lifespan of mouse models [173] . Therefore, novel drugs that selectively block SOAT1 enzyme can be developed to improve pancreatic cancer treatment. In addition, increased serum kynurenine/tryptophan ratio is associated with resistance to ICIs,indicating the possibility of combination immunotherapy targeting IDO [174] . However, early clinical trials of the combination of IDO inhibitors in PDAC were not effective, underscoring the multidimensional nature of immunosuppression in TME.
Epigenetic therapy
In addition to genetic mutations, epigenetic alterations have recently been recognized as important contributors to the development and progression of PDAC, as well as potential therapeutic targets. Epigenetic changes are heritable modifications of DNA chemistry or chromatin structure, including DNA methylation,histone acetylation, deacetylation and methylation, and miRNA changes, which affect gene expression without changing DNA sequence [175] . Despite the preliminary results obtainedinvitroandinvivostudies of drugs targeting epigenetic mechanisms, the results of clinical trials in patients with pancreatic cancer are disappointing and may be attributed to the lack of specificity of epigenetic drugs. About 80% of tumors overexpress DNA methyltransferase 1 (DNMT1) protein, including pancreatic cancer, and aberrant DNA methylation has been shown to play an important role in PDAC carcinogenesis, including promoting cancer cell proliferation, migration, and invasion, as well as inducing self-renewal of CSCs [176] . Early clinical trials of combination therapy involving DNMT1 inhibitor, including two classes of nucleoside and nonnucleoside, in PDAC patients are currently underway, which may improve the sensitivity of chemotherapy and ICIs. In the phase I/II solid tumor study, the combination of decitabine and immunotherapy regimen was tolerable with good efficacy [177] . The study by Nagaraju et al. [178] confirmed the inhibitory effect of curcumin and two curcumin analogues EF31 and UBS109 on DNMT1 expression and DNA methylation in pancreatic cancer, resulting in the growth inhibition of tumor cells, while these drugs increased the sensitivity of pancreatic cancer cells to 5-FU plus oxaliplatin. It was also found that curcumin and its analogues also inhibited the expression of miRNAs, including miR-101 and miR-199a, and decreased HMT EZH2 levels [179] . A phase II trial of curcumin in combination with gemcitabine for pancreatic cancer demonstrated that it had some biological activity and was well tolerated by patients [180] . Identification and characterization of novel DNMT1 inhibitors is a promising therapy for PDAC patients.
One of the key epigenetic signals regulating gene expression is acetylation and deacetylation of histone tails and other nonhistone internal lysine residues, and the enzymes responsible for this modification process include histone acetyltransferase (HAT),which transfers acetyl groups from acetyl-CoA to lysine residues,and histone deacetylase (HDAC), which reverses the above reaction [181] . At present, most of the possible therapies based on HAT inhibition focus on targeting the HAT CBP/p300 activity,but are only in the preclinical stage. With one exception, curcumin has been demonstrated to effectively inhibit HAT in cancer cells [182] and has shown early signs of clinical efficacy in patients with stage II PDAC [180] . HDAC inhibitors are well tolerated, and some promising agents have recently entered preclinical or clinical trials, including abexinostat, pracinostat, CUDC-101,belinostat (PXD-101), entinostat (MS-275) or panobinostat (LBH-589). Entinostat in combination with ICIs inhibited the activity of MDSCs and promoted immune responses and the sensitivity to ICIs in mouse models of PDAC [183] , which is being evaluated in clinical trials (NCT03250273). Additionally, histone methyl signatures may provoke long-term effects on cells by strong signaling through genetic expression patterns of certain genes, but the use of histone methyltransferase inhibitors (HMTIs) in pancreatic cancer is rare. Curcumin and its analogues mentioned above may reduce HMT EZH2 levels [179] and inhibit cell growth; Yuan et al. [184] discovered a novel HMTI called BRD4770 that mediated H3K9 methylation of HMTG9a and induced senescence in PANC-1 cells; In addition, gossypol (a natural product from cottonseed, a putative BH3 mimic) synergized with BRD770 to induce autophagy-related cell death in PANC-1 cells [185] . And the therapeutic value of these new drugsinvivois unknown. Both overexpression and down-regulation of non-coding RNAs (ncRNAs),mainly miRNAs, in pancreatic cancer are associated with poor survival and prognosis [186] . MiR-21 is upregulated in pancreatic cancer, associated with apoptosis inhibition, tumor proliferation, invasion, and chemoresistance [187]. Paik et al. [188] found that indole-3-carbinol (I3C) inhibited miR-21 expression and sensitized PANC-1 cells to gemcitabine; garcinol, a natural drug associated with anticancer activity, has been shown to alter the expression of several miRNAs including miR-21 and synergize with gemcitabine [189]and curcumin [190] to inhibit proliferation and induce apoptosis of pancreatic cancer cells. Recently, Bao et al. [191] found that metformin caused re-expression of miR-101, miR-200, let-7, and miR-26a, and treatment of pancreatic cancer cell lines with significantly reduced cell survival, colony formation ability, and increased sphere-disintegration. Currently, four phase II clinical trials of metformin in combination with other agents are recruiting patients (NCT01210911, NCT01167738, NCT016 6 6730, NCT01488552).In addition, strategies to directly modify miRNA levels also need to be developed, such as inhibition of overexpressed miRNAs with antisense oligonucleotides [192] , or restoration of let-7 levels in pancreatic cancer cells by plasmid-based synthesis of miRNAs or lentiviral induction to inhibit cell proliferation,KRASexpression,and MAPK activation [193] .
The crosstalk of genetic alterations with epigenetic changes not only causes neoplastic transformation, but also is likely to determine multiple characteristics of cancer phenotypes and their symptoms. While epigenetic drugs are regarded as targeted therapies, they often affect different pathways and tissues by exerting their effects throughout the genome in tumor and normal cells.Furthermore, these drugs that target epigenetic mechanisms may not act only through epigenetics, but involve multiple complex pathways. For example, HDAC inhibitors are not only involved in the deacetylation of histone tails, but a large number of other nonhistone proteins are also acetylated; 5-aza-dC exerts its demethylation activity throughout the genome and may therefore lead to the re-expression of hypermethylated tumor suppressor genes or activate oncogenes; furthermore, although new miRNAs and new targets of known miRNAs continue to be discovered, miRNA targeting not only affects cancerous pathways, but also affects other unnecessary normal pathways. Promising epigenetic-based therapies are currently being evaluated by different types of trials, and further studies are needed to establish all targets and downstream effects of epigenetic targeting in the future to provide a basis for understanding the causes of adverse effects, tumor specificity, and drug resistance, and ultimately to give the best medication regimen in terms of treatment options and safety in PDAC patients.
Therapy targeting autophagy
Autophagy has antitumor effects to maintain homeostasis during tumor initiation, however during tumor progression, it supports the catabolism of intracellular organelles, fuels tumor growth, and supports multiple types of tumor proliferation and survival [194] . Several autophagy inhibitors have been developed and tested in patients with a variety of malignancies, but with different results [195] . The autophagy inhibitor hydroxychloroquine failed to show activity in a phase II trial in patients with mPDAC [196] , but is currently being tested in combination with AG in a phase I/II trial (NCT01506973). In addition, MHC-I molecules have been found to selectively target lysosomal degradation through an autophagy-dependent mechanism that involves the autophagy cargo receptor NBR1 [197] , while autophagy inhibition restores MHC-I-level of PDAC and leads to improved antigen presentation,enhanced anti-tumor T cell responses, and reduced tumor growth in mice [197] . Thus, inhibition of autophagy by chloroquine could synergize with anti-PD-1 and anti-CTLA-4 antibodies to enhance antitumor immune responses, providing a new rationale for anti-PDAC therapeutic strategies.
Therapy targeting cancer stem cells
A preclinical study has identified a population of cells resistant to chemotherapy and with high tumorigenic potential, CSCs,within PDAC tumors, whose surface expression of CD44, CD24,and CD326 molecules supports self-renewal and therapeutic resistance [198] . However, the results of trials aimed at testing inhibitors that target differentiation pathways of CSCs, including Hedgehog, Wnt, and Notch, have mostly been negative [ 199 , 200 ].Napabucasin (BBI608) inhibitsSTAT3-driven gene transcription and CSCs formation, increasing the sensitivity of pancreatic cancer to chemotherapy pairs. A phase Ib/II trial of its combination with AG for mPDAC showed 78% disease control rate [201] , but one phase II trial (NCT02993731) was discontinued due to futility. Another international multicenter phase III RCT study is currently ongoing(NCT02993731). In addition, CSCs may have a range of metabolic and epigenetic alterations that represent a mechanism for their relative plasticity and response to local environmental changes, and strategies to target CSCs and epigenetic or metabolic processes appear attractive. These disappointing results may be related to the heterogeneity and plasticity of CSCs, and the use of single-cell sequencing help the understanding of tumor heterogeneity and the development of therapy targeting CSCs [202] . Despite setbacks, targeting CSCs is an important and active area of research in therapeutic resistance and disease aggressiveness.
The strategies of systemic therapy for pancreatic cancer are outlined in Fig. 1 and Table 1 .
Combination therapy in mPDAC
At present, monotherapy in PDAC cannot achieve satisfactory results, and more in-depth studies on gene targets, TME, and immune escape mechanisms are urgently needed to provide a new theoretical basis for reducing tolerance and developing new therapies. Combination treatment modalities that target many different mechanisms may have synergistic effects. For example, the combinations of gemcitabine and tumor vaccine, TNF-αand DCs vaccine, PD-1 antibody and CTLA-4 antibody, as well as therapeutic vaccine and ICIs may all produce better therapeutic effects, which have been validated in initial studies. Combination of OX40 agonist and anti-PD-1 antibody with neoantigen-targeted vaccine and STING agonist resulted in tumor regression and prolonged survival in mice with PDAC [203] . Notably, the results of several recent clinical trials in chemotherapy-based novel therapies have been disappointing, including PEGPH20, ibotinib, pegilodecakin (targeting pegylated recombinant IL-10), and the STAT3 inhibitor napabucasin.Nonetheless, the results of phase III studies of some new combination strategies may be reported in recent months and may establish new treatment modalities for this disease. However, toxicity, dose, and sequence are still key challenges in developing combination approaches, especially in agonists. In addition, the ideal therapy for combination therapy will also include the identification of biomarkers of response to cytotoxic agents, targeted therapies,and immunotherapies to rapidly assess treatment response. Major improvements in outcomes should be achieved in the next 5-10 years using novel agents that specifically target pathological signaling pathways and genetic alterations unique to individuals with PDAC. These novel agents, as a part of a combination approach,may need to act synergistically with conventional chemotherapy,targeted therapy, immunotherapy or other novel agents to prevent the emergence of early resistance.
Summary and prospect
PDAC is still an important and growing global health problem with high morbidity and mortality and a very low 5-year survival rate; it has few effective treatments which are being developed more slowly than that of other tumors. In the past decade,people have been continuously and widely committed to its exploration and research, whether in traditional chemotherapy, or targeted, immunotherapy and other new precision treatments. Although the results are not so satisfactory, they are also constantly revealing their mechanisms and fine-classifying their new molecular subtypes to achieve precise treatment and the study of precise molecular mechanisms. Recently, researchers have discovered a new method to classify pancreatic cancer subtypes and performed detailed genomic and transcriptome analysis, finally they distinguished five different pancreatic cancer subtypes based on molecular characteristics, which are basal-like A, basal-like B, classical A, classical B, and hybrid, each with unique molecular characteristics and can be used as targets for novel chemotherapies, biologics, and immunotherapies. This new classification system represents the most comprehensive analysis of molecular subtypes of pancreatic cancer to date. Substantial progress has been made in the understanding of the genetics and epigenetics of PDAC over the past decade, including the molecular basis of genomic and epigenomic heterogeneity affecting patient response to therapy and nat-
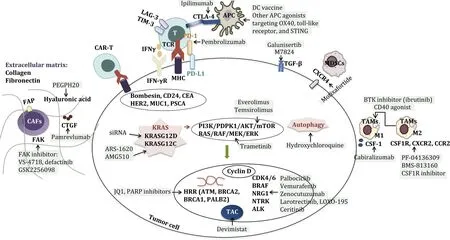
Fig. 1. Overview of systemic treatment strategies for pancreatic cancer. The figure summarizes the systemic therapeutic targets and corresponding drugs for pancreatic cancer, including treatment strategies for many aspects such as signaling pathways and gene mutations in tumor cells, immune cells and molecules in the extracellular environment, and extracellular matrix. Different targets are highlighted in bold black; each targeted drug is highlighted with a green background; the arrow indicates“targeting”. FAP: fibroblast activation protein; FAK: focal adhesion kinase; PEGPH20: pegylated recombinant human hyaluronidase PH20; CTGF: connective tissue growth factor; CAR-T: chimeric antigen receptor-T; CEA: carcinoembryonic antigen; CD24: cluster of differentiation 24; HER2: human epidermal growth factor receptor 2; PSCA:prostate stem cell antigen; KRAS: v-ki-ras2 kirsten rat sarcoma viral oncogene homolog; PARP: poly (ADP-ribose) polymerase; HRR: homologous recombination repair; ATM:ataxia telangiectasia-mutated gene; BRCA: breast cancer susceptibility gene; TAC: tricarboxylic acid cycle; ALK: anaplastic lymphoma kinase; CDK4/6: cyclin-dependent kinase 4/6; PI3K: phosphatidylinositol 3-kinase; PDPK1: 3-phosphoinositide dependent protein kinase 1; mTOR: mammalian target of rapamycin; MHC: major histocompatibility complex; LAG-3: lymphocyte activation gene-3; TIM-3: T cell immunoglobulin-3; TCR: T cell receptor; IFN- γ R: interferon- γ receptor; PD-L1: programmed death-ligand 1; PD-1: programmed cell death-1; CTLA-4: cytotoxic T-lymphocyte-associated protein-4; APC: antigen presenting cell; DC: dendritic cell; STING: stimulator of interferon genes; TGF- β: transforming growth factor- β; MDSCs: myeloid-derived suppressor cells; BTK: Bruton’s tyrosine kinase; TAMs:, tumor-associated macrophages; CSF-1R: colonystimulating factor 1 receptor; CXCR: CXC chemokine receptor; CCR2: C -C chemokine receptors 2; CSF-1: colony-stimulating factor 1.
ural history. With further development of various technologies, including comprehensive genomic analysis, single-cell analysis, and high-resolution imaging techniques, we expect to perform a wider range of genetic and molecular profiling to fully understand the interactions of these alterations within the TME ecosystem of PDAC and develop new target strategies. At present, based on the continuous understanding of molecular biology and molecular immunology, a variety of effective treatment strategies including traditional chemotherapy, targeted therapy, immunotherapy, microenvironment matrix modulators and epigenetic inhibitors have been developed, but due to the heterogeneous genes, molecular signaling pathways and complex immune TME of pancreatic cancer, a variety of treatment methods have failed. Effective combination strategies of different therapeutic agents may improve response to monotherapy and delay drug resistance. Furthermore, as more and more new potential therapeutic targets are discovered, a large number of studies are needed to validate the clinical application prospects of novel biomarkers in identifying effective therapeutic responses. Longitudinal monitoring and sampling of patients in clinical trials provide insight into the impact of various therapies on tumor subclonal evolution and heterogeneous TME components, which can help predict resistance mechanisms for immune escape, treatment failure, and proactively design combination targeting strategies. However, it must be recognized that even if multiple novel therapeutic intervention strategies using biomarkers/genetic identification are proposed, the clinical benefit of most PDAC patients still cannot be significantly improved; although we call for genetic testing, only a small proportion of the population can benefit from genetic test-based therapies.
Recently, a rather novel PDAC research field with its (great)therapeutic potential has been described. Investigators have proposed very strong arguments for adding the "transportome" to the"hallmark of pancreatic cancer" [204] . Over the past few decades,there has been substantial evidence that ion channels and transporters (ICTs) are significant in regulating physiological pancreatic duct cell function, and that dysregulation of ICTs is closely associated with widely accepted hallmarks of PDAC such as proliferation,invasion, differentiation, apoptosis, and metastasis. The study focused on the exocrine pancreas as a very active secretory gland,which significantly affects changes in ion transport systems upon malignant transformation, highlighting the diversity of ICT members (H+transporters, Ca2+, K+, Na+, and Cl-channels) and their functional impact in PDAC [204] . Therefore, selective treatment options that interfere with the key mechanisms of malignant progression by interfering with the function of the transporter (partially utilizing knowledge from neurobiology, cardiology, and other wellestablished pharmaceutical fields) will hopefully contribute to better clinical outcomes. Despite some new therapeutic ideas, PDAC is still an extremely deadly disease, and many new clinical trial results remain frustrating. Although most clinical trials on PDAC are still recruiting or designing and may not progress much in the next few years, it is still recommended to actively include all PDAC patients in future clinical trials for comprehensive evaluation. In the future, the occurrence, development, metastasis, drug resistance and other mechanisms of PDAC should be comprehensively described from the perspective of multi-omics; the molecular characteristics of pancreatic cancer genes, the code and metabolic mechanism of immunosuppressive TME should be deeply explored; alarge number of clinical studies have been carried out to gradually improve patient data collection and integration. Although it still faces the challenges of high tumor heterogeneity of pancreatic cancer, with the continuous exploration, from surgical local treatment to comprehensive medical management, the researchdiagnosis-management system of pancreatic cancer is improving,and moving towards precision, individualization, and finally continuously moving towards the goal of achieving high-quality longterm survival of patients.
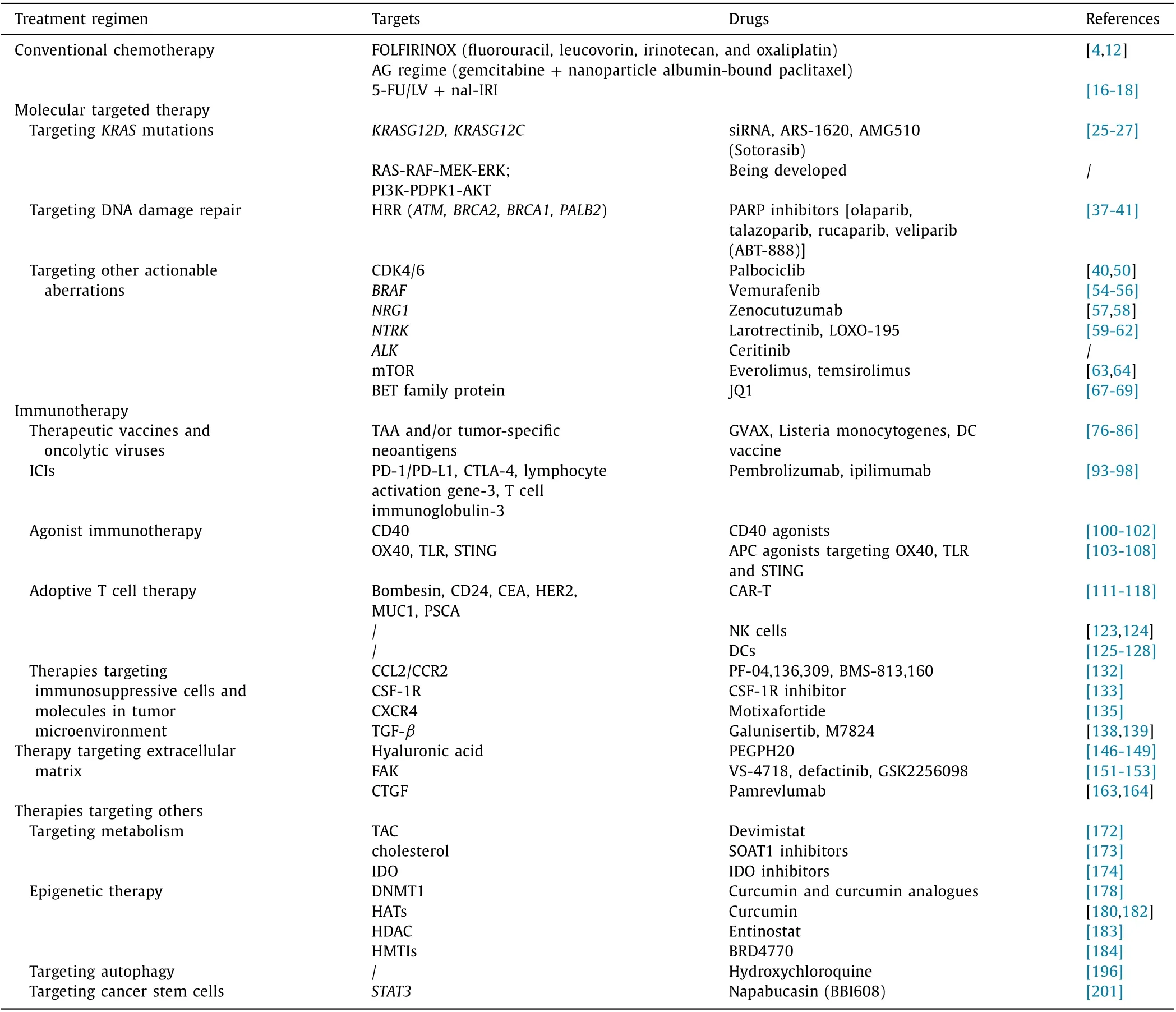
Table 1 Overview of systemic treatment strategies for pancreatic cancer.
Acknowledgments
None.
CRediT authorship contribution statement
Ri-Lan Bai : Resources, Supervision, Writing - original draft,Writing - review & editing. Nan-Ya Wang : Supervision, Writing -review & editing. Ling-Ling Zhao : Supervision, Writing - review &editing. Yong-Fei Zhang : Supervision, Writing - review & editing.Jiu-Wei Cui : Conceptualization; Funding acquisition, Resources, Supervision, Writing - review & editing.
Funding
The study was supported by grants from the National Key R&D Program of China (2016YFC1303800), Jilin Provincial Key Laboratory of Biological Therapy (20170622011JC), Jilin Provincial Science and Technology Department (20190303146SF), and Jilin Province Finance Department (2018SCZWSZX-010).
Ethical approval
Not needed.
Competing interest
No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
 Hepatobiliary & Pancreatic Diseases International2022年1期
Hepatobiliary & Pancreatic Diseases International2022年1期
- Hepatobiliary & Pancreatic Diseases International的其它文章
- Primary pancreatic lymphoma diagnosed by endoscopic ultrasound-guided fine needle biopsy
- A stable and reliable animal model for hepatocellular carcinoma with portal vein tumor thrombus
- Expert consensus on the application of the magnetic anchoring and traction technique in thoracoscopic and laparoscopic surgery
- SNHG16 promotes hepatocellular carcinoma development via activating ECM receptor interaction pathway
- Predictors of recurrent bile duct stone after clearance by endoscopic retrograde cholangiopancreatography: A case-control study
- Call for action: Increased healthcare utilization with growing use of percutaneous cholecystectomy tube over initial cholecystectomy in cirrhotics