
微反应器内连续制备拓扑结构聚合物的研究进展
2022-02-18 02:07向亮钟子豪苏远海
化工学报 2022年12期
向亮,钟子豪,苏远海
(上海交通大学化学化工学院化工系,教育部变革性分子前沿科学中心,上海 200240)
引 言
近年来,对聚合物的合成研究逐渐由传统高分子转变为拓扑结构聚合物的功能化定制。拓扑结构聚合物的制备过程既依赖于各种聚合技术的发展,即以(活性)自由基聚合、离子聚合、开环易位聚合等方法几乎能够定制任何可以想象的链结构,又借助于反应器操作工艺的完善,即通过模块化组合反应器的进料、反应、纯化、分离等多种工序,实现由单体到功能聚合物产品的规模化生产。微反应器作为一种重要的过程强化设备,能够实现可控的多相微尺度流动,强化聚合反应中的混合、传质和传热过程。相比于间歇操作,以微反应技术为代表的连续操作工艺在聚合物的分子量分布、分子结构和宏观形貌调控及聚合物生产效率等方面展现出优势。但微反应技术目前集中于对线形结构聚合物的定制,而对复杂拓扑结构的聚合物的设计及合成则报道较少,这有待相关研究者深入扩展。本文介绍了拓扑结构聚合物的结构性能、合成方法及微反应技术的理论基础背景,概括了微反应技术在聚合物的合成应用领域的优势,重点介绍了当前连续合成支链型和环状等复杂拓扑结构聚合物的研究进展,并展望了设计连续聚合过程中的混合传递、动力学模型结合、自动化控制及放大生产等未来主要研究方向,为微反应器制备拓扑结构聚合物的过程调控及产品性能优化提供重要的借鉴。
1 聚合物的拓扑结构
聚合物具有丰富的多层次结构且紧密相关,其中分子结构(即链结构)和分子的聚集态结构是决定其性能的最主要层次。小分子单体在聚合过程中被连接成特定链结构的大分子聚合物,形成原生聚集状态,可通过“信号”植入赋予高分子之间特定的相互作用力,进而在加工过程的外场作用下表现出所需的特定聚集态结构[1]。因此,对链结构进行有效设计及精密制造是提升高分子材料性能及附加值的关键[2]。近年来,对聚合物的合成研究逐渐由传统高分子转变为拓扑结构聚合物的功能化定制。这其中,含复杂拓扑链结构的聚合物的发展极大地丰富了高分子材料的种类与性能,比如含支链构型的聚合物具有独特的黏弹性,相比于同分子量的线形聚合物会表现出不同的溶液性能和热性能等。如何结合相关的聚合方法和反应器操作工艺实现对拓扑结构高分子精准、便利的合成,已成为高分子化工领域工作者的关注热点。
1.1 聚合物的链结构
如表1 所示,聚合物的链结构可分为紧密相关的三个层次。一级结构为官能团结构,即通过引入含官能团侧基的单体对主链骨架功能化,或使用含特殊官能团的引发剂、控制剂等对主链末端功能化,继而改变高分子材料的极性、溶解性、亲憎水性和生物相容性等本征特性。二级结构为序列结构或组成分布,即共聚物链上不同单体的排列顺序或所占分率,共聚物可按此分为四大类:无规、交替、嵌段、梯度。单体的共聚增加了聚合物的种类,拓宽了高分子材料的应用范围,而具有相同单体组成但含不同序列结构的聚合物会呈现出不同的性能。三级结构为拓扑结构,即聚合物链在空间的排布情况。除了最为常见的线形结构,拓扑结构还包括支链型、环状和多环形结构等。

表1 聚合物链结构分类[3]Table 1 Categories of polymer chain architectures[3]
拓扑结构是链结构的重要一环,并显著影响聚合物的相关性能。聚合物按拓扑结构主要分为线形、支链型和体型结构三大类。支链型聚合物含有连接到线形主链上的侧链,且侧链之间未通过化学键连接。体型聚合物的链段之间则通过化学键相互连接,进而形成具有三维网络的一个整体。不过,支链型和体型聚合物都可视为由线形基链贯穿于若干个支化点并按照一定的空间排布方式相互连结而形成的拓扑结构,前者还被细分为超支化、星形、接枝/梳状等[4]。当每条基链的一端可自由地连接到另一条链上且后者链上的单体单元具有同等被选择概率时,形成无规支化聚合物;当多条基链的一端与共同的核连接时,会形成星形聚合物;当每条基链的端基单元通过共价键相互连接而形成一条主链,这对应接枝/梳状聚合物;当超支化聚合物中每条线形基链上被平均分配至少一个支化点时,体系出现网络凝胶,这对应交联聚合物;当线性基链的首尾两端连接在一起时,这将形成环状聚合物。
1.2 拓扑结构聚合物的制备方法
各种新颖高效合成方法的涌现,使得聚合物拓扑结构的设计定制更为便捷,这极大地丰富了相关高性能高分子材料的品种。文献报道及生产实践中最常见的是线性聚合物,其可以通过多种聚合技术包括自由基聚合(FRP)、离子聚合、配位聚合及开环聚合等制备,在此不做赘述。本文中主要介绍支链型和环状的拓扑结构聚合物的制备方法。
1.2.1 支链型聚合物的制备方法 早期合成支链型聚合物常以ABx型[5]或A2+B3型[6]单体进行缩聚,主要用于超支化聚酯和聚氨酯的制备。但该合成路线要求单体含醇、异氰酸酯等功能基团,或者体系中含有特定的催化剂,此外存在缩聚动力学的平衡问题,这会降低反应速率,且单体转化率不高,产物易降解。后续出现以单烯和二烯的自由基共聚制备支化聚合物的方法。但共聚过程中,随着二烯参与交联反应程度的提高,聚合物从可溶的支化结构(溶胶)向不溶的交联结构(凝胶)转变。由于FRP过程中聚合物链是瞬间形成的,期间混杂交联反应的同时还伴随不可忽略的环化反应,这将使实际的凝胶点数值严重偏离理论预测值[7]。
1995 年Fréchet 等[8]提出的自缩合乙烯基聚合(SCVP)技术使支链型聚合物的合成研究取得重大突破。SCVP 基于自引发单体AB*,包含乙烯基A 和侧基B*, 其中B*能够引发聚合或转化成具有再引发能力的基团。AB*既作为引发剂又相当于聚合单元,通过彼此间的交替引发增长可进行支化均聚。不过SCVP 体系的自引发单体往往不能直接商业获取,而需在反应前进行烦琐的合成。
Sherrington 等[9-10]提出了一种新颖的Strathclyde合成方法。他们在FRP 过程中加入大量的硫醇作为链转移剂,通过增强链转移反应以降低线形基链的分子量,从而抑制聚合物链分子间的过度交联,避免形成凝胶。该方法通过单乙烯基单体与低含量的多乙烯基单体共聚得到长链支化聚合物,适用于一系列商业化的乙烯基单体,包括苯乙烯(St)[11-12]、甲基丙烯酸甲酯(MMA)[9,13]、乙酸乙烯酯[14]等。不过由于体系中引入大量链转移剂,聚合产物的分子量分布往往较宽。
20 世纪90 年代兴起的可逆失活自由基聚合(RDRP)技术,为支链型聚合物的制备和复杂结构的精确控制提供了新的技术依托。目前应用最广的RDRP 技术主要有三种:氮氧稳定自由基聚合(NMP)、原子转移自由基聚合(ATRP)和可逆加成断裂链转移自由基聚合(RAFT)。RDRP 体系中通过加入一种钝化链增长自由基的化合物,建立活性种与休眠种的可逆平衡,显著降低体系内自由基浓度,进而减缓链增长速率,同时抑制自由基的双基终止。将RDRP 技术用于单烯与二烯的共聚体系时,较低的自由基浓度使聚合物链具有充足的舒展和扩散时间,因此由二烯提供的悬挂双键能与其他增长链自由基充分地接触,这有利于生成均匀的支化/交联结构,减少微凝胶的形成[15]。
联用RAFT 与SCVP 技术,即将单烯与二烯、RAFT 试剂一锅法共聚或将含有RAFT 官能团的单烯单体进行均聚,可制得超支化聚合物。Wang 等[16]采用半连续操作,通过不同加料策略,以单烯/二烯的RAFT 共聚定制了超支化聚丙烯酰胺(b-PAM)。Liang 等[17]从RAFT 聚合的基元反应出发,建立了与实验结果匹配的单烯/二烯支化共聚动力学模型,可用于指导超支化PMMA 和聚丙烯酰胺-海藻酸钠双网络凝胶的合成。
星形聚合物是一类含有至少三条链(臂)且各条链无主、支链的区分,以化学键连接于同一点(核)而形成的支链型聚合物,其通常采用“臂先”(arm-first)与“核先”(core-first)两种策略来制备。在“臂先”法中,首先使单体转化为线形的聚合物臂,随后与交联剂发生偶合反应形成星形分子。“核先”法常采用多官能团的引发剂作为核,通过调整交联核的官能团含量以精确控制星形聚合物的臂数目。Wang 等[18]详细比较了采用不同二烯进料模式以RAFT 聚合制备星形聚丙烯酰胺(s-PAM)的结果,发现“核先”法产物的二烯利用效率更高,但分子量调控困难,而“臂先”法更适合对高分子量、多臂数的s-PAM的定制。
接枝聚合物由众多的线形侧链接枝于一个中心骨架构建而成。当接枝密度足够高时,侧链之间相互排斥使得主链被迫伸展,整个聚合物的构象类似于梳子,此时被称为梳状聚合物,其制备策略包括:(1)收敛法(graft to),通过特定官能团的反应将侧链和预聚物主链连接在一起;(2)发散法(graft from),在先合成的主链上的活性位点再引发单体聚合得到侧链;(3)大分子单体法(graft through),以末端含双键或其他反应基团的大分子单体进行聚合。接枝聚合物的分步制备往往采用多种合成方法的组合。如Perrier 等[19]采用graft from 策略,先对N-(2-羟乙基)丙烯酰胺进行RAFT聚合得到主链,后使单体单元的羟基与小分子RAFT 的羧基发生Steglich 酯化反应,从而侧链“活性”链增长形成丙烯酸酯类的嵌段共聚物,最终得到复杂的瓶刷状构型聚合物。采用相同策略,Jia 等[20]先以RAFT 制备含丙烯酸丁酯(BA)及螺吡喃单元的共聚主链,后以ATRP 技术接枝上PMMA 侧链。他们通过调控接枝共聚物中PMMA 分散相的尺寸,开发了一种基于荧光共振能量转移技术的力致变色材料。
1.2.2 环状聚合物的制备方法 环状聚合物是一类具有特殊拓扑结构的聚合物,其每个重复单元具有等同性。环状聚合物可划分为单环或多环聚合物,而单环聚合物是各种环状结构聚合物研究和构建的基础,合成方法主要包括扩环法和关环法。扩环法中,通过引发剂活化环形单体,再进行扩链增长实现聚合。关环法中,则是首先合成线形聚合物,然后使端基功能化作为线形前体,继而通过偶联反应闭环(包括分子内关环与分子间关环)形成环状聚合物[21]。如Pan等[22]采用扩环法,先设计一种环状二硫代酯RAFT 试剂,调控丙烯酸甲酯(MA)的低温辐射聚合后制备分子量在8 kg/mol 左右的高纯度环化PMA,再对N-异丙基酰胺(NIPAM)进行扩链聚合,最终得到环状嵌段共聚物PMA-b-NIPAM。Huang 等[23]则选用分子内关环的路径,先以阴离子聚合制备端基为羟基的线形聚苯乙烯,然后以羟基为引发位点,通过阴离子开环聚合在线形链的两端接枝上聚环氧乙烷,继续将聚环氧乙烷链端的羟基修饰为炔基,最后以Glaser 偶合反应闭环得到环状嵌段聚合物。
1.3 拓扑结构聚合物的性能及应用
拓扑聚合物的性能与其结构紧密相关。支链型聚合物常表现出三维椭球状的空间构型,分子链缠结作用较小,较难结晶。高度支化的分子外围富集了大量的活性基团,它们大量堆积使聚合物中心形成了密闭式的空腔结构,同时这些功能基团为进一步直接或间接的改性提供了有利条件。通过控制功能基团的数量和类型,可以实现对支链型聚合物溶解性、反应性、界面自组装、表面吸附以及电化学和光学性质等多领域的调控。典型的如星形聚合物,由线形链和支化点相应构成的臂和核因其不同特性,使聚合物能够在本体、溶液等相界面上发生相分离,随后形成的聚集体(如胶束)中的隔离空间可以提供特定的化学环境以储存各种小分子。在星形聚合物的致密链段中引入对热、光、pH 等响应的单体单元,可定制灵敏的智能材料等。相比于同等分子量的线形聚合物,支链型聚合物具有更低的玻璃化转变温度和熔融温度、更好的溶解性、更强的负载能力,以及更密集的功能基团等特性[9,24-26],这些独特的优势使支链型聚合物在乳化、催化、光子学、药物输送、复合材料等领域具有广泛的应用[27-29](图1)。

图1 支链型聚合物的结构性能关系及相关应用[29]Fig.1 Structure-property relationships of branched polymers and related applications[29]
类似地,环状聚合物相比于线形聚合物没有链末端,因此其物理、化学性质不受“端基效应”影响。与线形聚合物相比,环状聚合物具有较高的密度和玻璃化转变温度,较低的特性黏度和较小的流体力学体积[30]。在荧光性质上,相同分子量的大环聚合物的荧光强度更高,荧光寿命更长[31]。在结晶性能上,环状聚合物的结晶速率更快[32]。在溶液中,环状聚合物具有更低的临界溶液温度和更高的折射率[33]。在生物应用方面,环状聚合物应用于基因载体表现出更低的细胞毒性和更高的基因搭载效率[34]。
2 微反应技术对聚合物拓扑结构的调控
2.1 微反应器及其特性
微反应技术兴起于20 世纪90 年代。狭义上,微反应器是指特征尺度在微米至毫米之间的用于进行化学合成的管式、板式或芯片式的反应装置;而广义上,它是包含混合、反应、换热、控制、分离、化学分析等各种微型化工设备的高度集成的微反应系统[35]。微反应技术的应用促进了化工设备小型化和化工过程集约化,大幅提升了资源利用率,从根本上解决了传统间歇工艺的效率低、能耗高及安全性能差等技术难题。
微反应器中,流体流动、传质、传热和反应这四种基本的化工过程被耦合在微小的受限空间,因此,微反应器在流体力学、混合传递、反应和分离等方面主要表现出如下优势[36-38]:
(1)扩散距离短,速度、温度、浓度、压力等物理量梯度大,对应的化工过程中传质传热的推动力较大,因此具有较高的混合效率和热质传递速率;
(2)通过成熟的微加工技术可将混合、反应、换热、分离等多个单元操作与相匹配的传感、阀门等器件集成到微反应器中,因此整个装置体系高度集成化;
(3)可通过改变微通道的长度与流量设计停留时间,调节多种反应参数如温度、压力以优化产物的时空收率和选择性,因此能实现对反应条件的精确控制;
(4)反应持液量少,减少有毒物质的生成,同时大比表面(比表面积达5000~50000 m2/m3)使得强放热反应体系产生的热量能被快速移除,避免“飞温”,因此微反应器的反应安全性能高;
(5)可通过并行增加反应管的数量实现微反应器的快速放大,而不需要进行传统的尺寸放大,这可以缩短工业放大工序,使实际生产更为灵活,因此操作易于放大[39-40]。
2.2 微反应器在聚合物合成的应用
聚合物的合成是微反应技术应用的一个重要领域。最早报道微反应器内聚合过程可追溯到20世纪60 年代初,Szwarc 等[41]构建了一种连续毛细管装置,能实现单体和含活性基团聚合物的高效混合及在管末端的反应快速终止,在室温及0.1~0.8 s 的反应时间条件下研究了St 的阴离子聚合动力学,并制备了分子量分布较窄的聚苯乙烯(PSt)。这一思路正是聚合过程强化的关键,即利用微反应器操作工艺,应用于常见的聚合反应包括自由基聚合、阴/阳离子聚合、缩聚等强放热快速反应过程,控制产物的分子量结构。随着微反应技术的发展,以其调控聚合物的链结构、聚集态结构和宏观形貌也逐渐成为微尺度聚合研究的热点。
聚合物的分子量控制是聚合物合成中最基本的研究课题之一。链式聚合(自由基聚合和离子聚合)的分子量分布主要受体系温度、反应物浓度分布的影响,逐步聚合(缩聚)的分子量分布与反应器内的停留时间分布有关。微反应器良好的混合、热质传递性能及接近平推流的反应器操作特性,使其对链式聚合和逐步聚合过程都具有较好的分子量调控。Yoshida 等[42]详细研究了五种单体在微反应器中的FRP 动力学特性,发现相比于间歇聚合,高放热的聚合体系包括聚甲基丙烯酸苄酯、聚甲基丙烯酸甲酯及聚丙烯酸丁酯的分子量分散系数(Đ)显著降低,如聚丙烯酸丁酯的Đ可从10 左右降至3.16,而对于低放热的体系如PSt 和聚苯甲酸乙烯酯,则没有这种明显的影响。Theato 等[43]报道了一种从三烷氧基硅烷到聚倍半硅氧烷的连续缩聚过程。由于微反应器内的混合效率高,反应时间可以从间歇操作的3 h 缩短到0.22~2.64 min 范围内,聚合物的分子量偏差从50%降低到3.4%,即微反应器操作可实现更快的反应速率和更高的聚合物产率。
在聚合物的链结构控制方面,微反应器模块化的组合方式为含功能端基、不同序列组成及复杂拓扑结构的高分子的可控设计及制备创造了有利条件。Zha 等[44]发现在微反应器中导入惰性气体可极大地改善微观混合效果,采用氮气-高分子溶液两相流强化的方法制备磺化PSt。Nagaki 等[45]使用三级串联的微反应器实现了St 和丙烯酸酯类单体的嵌段共聚。不同的微反应模块可选择独立的温度控制单元,他们对此进行工艺优化,在分段温度控制下实现了对PSt 的端基改性及三嵌段共聚物的连续合成。
在聚合物的形貌控制方面,以微反应器精确调控内部的多相流型及液滴的体积、数量和组成,通过多级串联等操作工艺,可以构建单分散液滴、单分散多重乳液、Janus 液滴等分散相流体,而液滴内的单体经聚合、光固化交联后,可形成具有特殊宏观形貌的单分散微球、多重包覆颗粒、多芯微胶囊等。Xu 等[46]以多级微流控装置制备的稳定O/W/O双重乳液为模板,通过原位紫外光引发FRP 后得到核壳微粒(图2)。由于水凝胶外壳层的保护,在多个分隔内核中的量子点不易被泄漏且荧光特征波长互不影响,荧光性能在局域空间内增强,具有很好的医学成像应用。Song 等[47]采用多股进料,在微反应器内以弹状流或液滴流的操作实现了对聚吡咯/二氧化锰(PPy/MnO2)核壳结构复合物材料的可控制备,产物用于超级电容器的电极活性材料,比块状形貌的PPy/ MnO2复合物或MnO2表现出更大的比电容量。
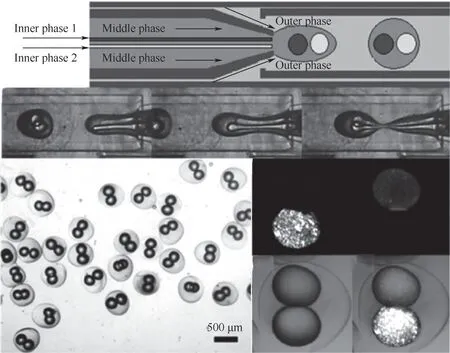
图2 利用微反应器制备多色量子点编码的聚合物核壳微粒[46]Fig.2 Preparation of multicolor quantum-dot-encoded core-shell microparticles in a microfluidic platform[46]
2.3 微反应器中的RDRP
FRP因其反应条件温和、适用于多种聚合方式、单体适用面广等优点而被广泛应用于工业生产。据统计目前市面60%以上的大宗聚合物是通过FRP 方式生产的。RDRP 技术在继承这些特点的同时,其强大的分子设计能力在新材料结构性能的设计定制方面引起了研究者们的广大关注。而微反应器在聚合物的分子量分布控制、链结构和宏观形貌调控等方面具有优势。因此,将RDRP 与连续流微反应技术相结合,可逐步拓展RDRP 聚合技术的应用领域。
如对于NMP 聚合,其聚合温度往往超过100℃且体系是放热反应,而微反应器良好的传热特性可以使产物的结构调控优于间歇操作。Serra 等[48]进行微反应器中的NMP 均相聚合,发现摩尔放热量低的St 聚合与间歇釜的操作结果相似,而放热量高的BA 的聚合速率和分子量调控显著优于间歇聚合。Studer 等[49-50]在St和MMA 的聚合体系中得到类似的结论,他们还合成两嵌段共聚物,发现产物性质受微混合器的几何构型(如通道数、特征尺寸等)影响。在ATRP 聚合领域,Zhu 等[51]报道相关连续流操作的应用。他们将硅胶负载的CuBr-HMTETA 配体填充于铜管中,进行MMA 均聚和MMA-BMA 的两嵌段共聚。该微通道反应器可以长时间运行并保持较好的催化活性,产物无须除铜处理。在RAFT聚合方面,Hornung 等[52]开展了乙酸乙烯酯类、丙烯酰胺类、丙烯酸酯类等多种单体的连续溶液聚合,均获得窄分子量分布的均聚物及两嵌段共聚物。他们在微反应器系统中集成在线脱气、聚合、产物沉淀分离、RAFT 端基脱除等工序,开发了由单体到纯净聚合物的一体化制备工艺[53]。Cheng 等[54]在不锈钢微管中进行了MMA 的自稳定无皂乳液聚合和水溶性单体的两嵌段共聚,体系均表现出良好的活性特征(Đ≤1.2)。他们调整串级进料的单体分别为亲水性和疏水性,可制备两亲性嵌段共聚物,并发现在连续流动的聚合诱导自组装过程中,聚合物胶束的粒径受混合装置(T 型三通或静态混合器)的选型影响[55-56]。他们还在石英材质的微管中以可见光的RAFT 聚合制备水溶性聚合物,除考察光源波长的影响外,发现该体系在实现单体高转化率的同时可简化除氧操作[57]。
值得一提的是,部分RDRP 热聚体系的反应速率较慢,放热不显著,并不会明显受益于微反应器优良的传热传质特点,但通过对微反应器多种模块组合等操作方式的改进,可提高聚合过程的能量利用效率,快速优化反应条件,推进产品放大生产的研发进程。
3 微反应器对拓扑结构聚合物的调控
3.1 微反应器对线形聚合物的调控
线形聚合物的空间排布最为简单,这也是当前微反应技术应用于聚合物合成中报道最多的拓扑结构(三级结构)。进一步细分至对线形聚合物的二级结构的调控,已在微反应器中连续制备出包括多嵌段、梯度和单分子序列寡聚物等不同序列结构的聚合产品,其合成精度、效率往往高于间歇过程。
如对于多嵌段(嵌段数≥3)共聚物,Boyer 等[58]利用曙红Y 和三乙醇胺的光催化体系,串联多个微反应器,基于多种丙烯酰胺、丙烯酸酯类单体,在10 min 的绿光光照条件下合成了PDMAA-b-PDMAAb-PDMAA 三嵌段共聚物,分子量可达1 万,产量达到300 g/d。Junkers 等[59]通过串级微反应器中丙烯酸酯类单体的顺次进料,以100℃高温加快每段单体RAFT 热聚的反应速率,在40 min 的总停留时间内得到Đ≤1.47的四嵌段共聚物,并进行了连续运行26 h获得150 g产物的放大生产。Perrier等[60]设计一种环管反应装置,采用各嵌段增长过程中的单体注入、循环流动、产物收集的操作工艺,以RAFT 热聚制备得到含六嵌段的丙烯酰胺类共聚物。局限于对多嵌段聚合物的各段活性控制,这些报道中的嵌段聚合物链节数目较低,数均分子量在1 万以内。对此,有望通过微反应器中的液滴流、气液两相流等非均相操作提高聚合产品的分子量[61]。Mastan等[62]针对环管微反应器中苯乙烯/丁二烯的阴离子共聚体系,模拟了连续制备含多嵌段数共聚物的聚合过程并分析产物的链长及组成分布,产物分子量可达10万以上,有望用于热塑性弹性体。
梯度聚合物在以间歇操作制备时,对单体的适用范围、梯度趋势可调区域有较大限制,而以半连续加料操作合成时,易遇到分子量分布宽、链段活性低的问题。对此,Zhou 等[63]开发了计算机辅助的流动化学活性梯度共聚,即在微反应器中,基于不同单体在液滴与载流体的溶解度差异,使竞聚率相近或差别较大的单体均能以RAFT 聚合得到梯度共聚物(图3),该方案具有产物分子量分布窄、可程序合成等优点。

图3 (a)计算机辅助的液滴式光照流动聚合装置;(b)基于单体扩散原理的光控连续制备梯度共聚物[64]Fig.3 (a) Computer-aided droplet-flow setup for photopolymerization; (b) Process for the synthesis of gradient copolymers via the droplet-flow photopolymerization based on the monomer diffusion[64]
对于单分子序列寡聚物,Leibfarth 等[65-66]提出了一种流动-迭代指数增长(flow-IEG)的操作工艺。他们在微反应器系统中集成了单体的选择性脱保护、在线纯化、点击反应等工序,解决了间歇IEG 增长中纯化步骤烦琐的问题,而且能够规模化制备单分散聚合物。Xu 等[67]在光照条件下通过连续和交替插入不同单体,精确合成了包括五个单体单元的低聚物。该微反应器的五步迭代操作产率达到59%, 能够在较短的生产时间内(数天)实现克级产物的制备,为合成序列精确的寡聚物提供了新合成技术。
3.2 微反应器对支链型聚合物的调控
相比于线形聚合物,支链型聚合物的连续合成报道较少,以下详细列举已知的微反应器对各种支链型聚合物的调控报道。
对于超支化聚合物,Isaure 等[68]在2006 年报道了通过平行微通道装置合成水溶性的超支化聚二甲基丙烯酰胺。与烧瓶中的间歇合成相比,连续操作的产率更高,且两者产物略有不同,这可能是因为微反应器的密封性更好,有效排除了体系中微量的O2。Liu 等[69]使用含交叉指型微混合器的微反应器系统,采用多步缩聚法,在室温(而非间歇操作要求的0℃低温)条件下,将每一步的反应时间从几小时缩短至几秒钟。Wilms 等[70]在微芯片装置中以阴离子开环聚合制备单分散的超支化聚缩水甘油。该放热体系受益于微反应器高效的传热传质性能,避免了间歇操作中耗时的单体慢滴加工序,聚合时间显著缩短。Serra 等[71]利用ATRP SCVP,在连续流中合成支化PMMA,其相比间歇产物具有更高的支化度(图4)。他们对螺旋微反应器加以改进,使流动方向突变的“拐角”增多,可削弱在高转化率时因黏度增加而导致的扩散限制,极大地增强混合程度,进而提高聚合产物的支化效率。Junkers 等[72]在微反应器中以光引发自由基本体聚合制备支化聚丙烯酸丁酯,20 min内单体几乎完全转化,通过24 h的连续放大可稳定生产300 g 支化产物。Xiang 等[73]采用Strathclyde 方法,在链转移剂十二硫醇的调控下以丙烯酸丁酯和二乙烯基苯共聚,在微反应器中得到了不同支化程度的超支化聚合物。他们详细考察反应配方工艺的影响,发现微反应器更好的传递特性有利于提高产物的支化效率并避免局部热点下的副反应。此外他们还探索了更为环保的细乳液连续制备超支化聚合物的路径,相比于溶液聚合,细乳液体系的反应速率更快,产物的支化度更高。Advincula 等[74]利用RAFT SCVP,在室温透气环境以水/甲醇为溶剂,连续合成水系的超支化聚(聚乙二醇甲基醚)丙烯酸酯,再以热解气相色谱-质谱联用等手段详细表征了产物的支化结构。

图4 以间歇和微反应操作得到的支化聚合物比较[71]Fig.4 Schematic comparison of branched polymer synthesis in batch and microreactors[71]
对于星形聚合物,Junkers 等[75]在微反应器中结合光控ATRP,以多官能团的环糊精为引发剂核,按“核先”策略得到4、6 及21 条臂的星形聚合物,且每种臂进行多次扩链反应,最终产物具有对pH 的刺激响应性。Yin[76]以类似方案,先将SiO2微球与含Br的ATRP 引发剂通过化学键连接后填充到色谱柱上,再通过SiO2核上的多个活性位点原位链增长,连续制得含有N-异丙基丙烯酰胺和对氯苯乙烯单元的嵌段共聚物臂的星形聚合物,产物具有设计的表面接触角及染料负载功能。“核先”法中引发剂上的官能团数目限制了臂的数目,而且线形臂的组成及分布较难检测,对此Junkers 等[77]发展紫外光照下的“臂先”法,在20 min 的停留时间内合成了臂数约为30、重均分子量18万的PMA-PBA 杂臂星形聚合物。Xiang 等[78-79]搭建了一种毛细管串级微反应器系统,基于对微反应器中第一步形成线形臂和第二步形成星形大分子的动力学研究,优化反应配方及操作工艺,以RAFT“臂先”法连续制备了一系列满足设计臂组成的星形聚合物(图5)。考虑到星形聚合物的功能基团多、组成便于调控的特点,他们制备含有亲水/疏水臂或温度/pH 响应功能臂的星形聚合物,产物可以直接用于乳化剂或作为金属纳米粒子催化剂的载体。

图5 以串级微反应器连续制备刺激响应型星形聚合物,产物用于Au催化剂的载体[78]Fig.5 Schematic overview of the cascade microreactor system for the continuous synthesis of stimuli-responsive star polymers, and the products were used to support the Au catalyst[78]
对于接枝/刷状聚合物,Guironnet 等[80]先通过计算机程序控制毛细管中连续操作的停留时间,以开环聚合制备不同链长的大单体。然后大单体半连续地进入烧瓶中,发生开环易位聚合(ROMP)反应并以graft through 方式接枝到线形主链上。他们结合实验与分子模拟,发现由电镜观察到的聚合物宏观形貌近似于所设计的“瓶刷”结构。Boyer 等[81]在反应体系中引入两种具有不同特征波长和链转移能力的RAFT 试剂,通过调控微反应器两段反应部分的外部光源,无须中间体除杂工序等,可快速切换生产含接枝或超支化构型的聚合物产品(图6)。Guo 等[82]利用串级微反应器,第一步通过负载酶的固定床大大加快己内酯的开环聚合反应效率,第二步以graft through 的策略使降冰片烯基团发生ROMP反应,最终得到瓶刷状聚合物。

图6 微反应器内通过切换光源选择性制备超支化或接枝聚合物[81]Fig.6 Selective production of hyperbranched or grafted polymers by switching light sources in microreactors[81]
3.3 微反应器对其他复杂拓扑结构聚合物的调控
对于具有其他复杂拓扑结构聚合物合成的调控,有关微反应技术的应用主要集中于环状聚合物。如图7 所示,Zhang 等[83]采用关环法,即先以RAFT 聚合制备线形的PSt,然后通过微反应器进行UV 光照,在极稀溶液条件下,使PSt 一端的邻醌二甲烷转化为共轭双键,再与另一端的二硫酯发生Diels-Alder反应,从而制备得到环状的PSt。该连续操作得益于光照的均匀性,比间歇操作的产量更高。Baeten 等[84]在Zhang 等[83]方案上添加了循环装置,使未充分转化的原料返至微反应器入口并继续参与反应。这种环管型设计在达到直管反应器相同的生产能力时,反应器体积可降至直管的1/43。Barner-Kowollik 等[85]通过切换不同光源的波长使两种光反应互相正交,即功能大单体在350 nm 光源下发生链增长,而在410 nm 时发生[2+2]环化反应,从而在串级微反应器中实现了环状聚合物的连续合成。Shen 等[86]将线形聚合物(聚环氧乙烷或聚3-己基噻吩)和催化剂通过微混合器发生Glaser 偶联成环。他们发现反应速率、环化效率主要和两相的相对流速有关,表明反应受扩散控制,且添加回流泵装置能使催化剂相循环利用,从而提高了原料的转化效率。

图7 以间歇或连续方式制备环状聚合物[83]Fig.7 Preparation of cyclic polymers under batch and continuous-flow reaction conditions[83]
4 微反应器调控复杂拓扑结构聚合物的难点与改进方案
目前,通过微反应器连续调控复杂拓扑结构聚合物的研究仍处于兴起阶段,主要存在以下难点。
(1)复杂拓扑结构聚合物在合成过程中的溶液黏度可能存在较大变化。例如在相同分子量下,支链型聚合物的特性黏度低于线形聚合物,这在理论上适合于连续操作。但是由于支化过程存在凝胶点的转变,即此时聚合物的分子量急剧增加,体系黏度骤升,使得微反应器内的阻力和压降增大而造成通道堵塞。此外,微反应器的内壁或接口处附着交联聚合物后,很难通过溶剂冲洗去除,可能导致微反应装置存在难以重复使用的问题。
(2)复杂拓扑结构聚合物的合成路线较为复杂,如对于星形聚合物,不管按照“核先”还是“臂先”法,单体与交联剂都必须顺次加入、多步聚合;对于环状聚合物,无论是按照扩环法或是关环法,都要对聚合物链段进行相应的修饰,逐步聚合,并对每步产物分离纯化[33]。如何将这种复杂的聚合物合成过程在连续装置中实施,这对微反应器的设计与操作工艺提出挑战。
(3)复杂拓扑结构聚合物的结构表征有待改进。这既受到高分子检测手段的制约,也源于连续操作过程中很难对聚合动力学进行实时在线监控。而收集微反应器内的不同空间位置处产物的状态信息,对了解聚合进程及快速优化反应条件等起到重要的作用。
对此有如下改进方案。
(1)为了避免支化过程发生凝胶,需对反应配方进行工艺优化,如针对FRP 体系选用具有高链转移能力的硫醇或RAFT 试剂等。此外,需要控制反应进程以避免达到凝胶点转化率,这可以通过相关的动力学模型预测[15,87]。
(2)针对多步操作,可以设计微反应器的串、并联操作,并优化多个流股中的流量、反应时间,避免可能的返混等不利影响[78-79]。通过微反应器的外场强化,如以光热耦合手段加快反应速率,提高反应物的转化率,从而减少聚合过程中的中间体纯化工序[88]。
(3)针对复杂拓扑结构聚合物的表征问题,在合成研究中应选用研究成熟的单体体系,如苯乙烯、(甲基)丙烯酸酯类单体,并结合多种检测手段如凝胶渗透色谱,多维核磁共振及在线红外、紫外等检测和分析产物结构[26]。此外,借助与实际情形相匹配的动力学模型,可通过模拟实时预测实验体系难获取的关键动力学指标,这有利于加深研究者对聚合反应过程的认识并指导工艺优化[89-90]。
5 结论与展望
拓扑结构作为聚合物多层次链结构中的重要一环,极大地影响了高分子材料的物理化学性质,赋予其全新的应用潜能。聚合物的合成制备依托于聚合方法、反应器及操作工艺。以RDRP 技术等为代表的聚合方法在聚合物链结构调控方面蕴含着巨大的分子设计潜力。微反应技术也在提高聚合产品收率、优化反应条件及放大生产上具有突出的优势。基于微反应器的连续操作工艺能对聚合物的分子量分布、链结构和宏观形貌进行多方面的有效调控,然而相比于线形聚合物,支链型等复杂拓扑结构聚合物的连续合成在近些年才兴起。考虑到拓扑结构聚合物的应用潜力,将微反应器用于聚合物的链结构调控及连续生产将具有很强的前瞻性和很高的实践意义。
迄今为止, 国内外学术界已陆续报道了利用微反应器技术对超支化、星形、接枝、环状等拓扑结构聚合物的连续合成,且取得了较好的成果。然而在基础研究方面,针对毛细管微反应器内支化过程如何受到传质、扩散的影响,如何优化反应配方及操作工艺以避免体系发生凝胶,以及如何充分认识连续微反应装置中各处的反应物性质、动力学特征,这有待高分子化工研究者们深入探索。这既需要了解高分子反应机理并剖析相关的聚合过程,还要求掌握化工单元操作以深刻探究反应与传递之间的关系。此外,可考虑采用自动化在线检测平台对聚合产物性质实时分析,以及利用合适的数学模型对连续过程进行预测及匹配等。在工程放大方面,应用于聚合物并行放大生产过程中的低能量损失、流动分布均匀的流体分布器结构仍需开发及优化,对装置长时间运行时体系潜在的凝胶风险需做及时的预防。在产品应用方面,应把握聚合物结构与性能的关系,以产品性能为导向去设计聚合物的链结构,进而耦合相关的微反应器操作,实现对期望产物的连续制备,并扩展到乳化、催化、控释、光电材料等新兴领域,从而推进微反应技术与功能高分子材料研究的产业化应用。
猜你喜欢
纺织科学研究(2021年7期)2021-08-14
铁道建筑技术(2021年3期)2021-07-21
西部交通科技(2021年9期)2021-01-11
铁道建筑技术(2020年11期)2020-05-22
农业环境科学学报(2017年2期)2017-03-20
现代检验医学杂志(2016年1期)2016-11-12
中国塑料(2015年1期)2015-10-14
中国生化药物杂志(2015年4期)2015-07-07
北京航空航天大学学报(2014年1期)2014-12-19
中国机械工程(2012年15期)2012-07-25
