
多尺度相改性碳泡沫复合材料的制备及性能
2022-02-16 06:33:02侯林伟贺辛亥赵建伟苏秉尧袁亚蓉
纺织高校基础科学学报 2022年4期
侯林伟,王 斌,贺辛亥,赵建伟,苏秉尧,袁亚蓉
(1.西安工程大学 材料工程学院,陕西 西安 710048;2.西安工程大学 西安市纺织复合材料重点实验室,陕西 西安 710048)
0 引 言
碳泡沫[1-2]是一种由泡孔相互连接而成的三维网状多孔材料,因其密度小、孔隙率高、耐腐蚀等显著特点引起人们的广泛关注[3-4]。除了具备传统碳材料的一些特性之外,碳泡沫特殊的力学性能和优良的耐热性,可与金属或非金属结合,从而得到高性能的结构材料[5-6]。
为进一步提高其性能,相关学者研究了碳泡沫材料的改性。宋佳音等将蒙脱土引入碳泡沫,通过改变泡沫孔径,提高了碳泡沫的隔热性能与阻燃性能[7]。康宇鹏等在碳泡沫材料中原位合成碳纳米线,一定程度上改善了其力学性能[8]。另有学者通过模板法制备性能优良的碳泡沫材料。吕龙飞以密胺海绵为模板,制备了吸波性能良好的碳泡沫材料,在一定程度上缓解了周围环境的电磁污染[9]。
近年来,一些学者以聚合物前驱体为模板,经高温热处理得到碳泡沫材料。FARHAN以酚醛树脂为前驱体,聚氨酯为模板,纳米沥青颗粒为增强相制备了压缩性能较优的碳泡沫复合材料[20]。LI通过将酚醛树脂浆料浸渍在多孔聚氨酯表面,经碳化-化学气相沉积(CVD),合成了具有优良电磁屏蔽性能的轻质热解碳泡沫[21]。因此,根据应用需求的不同,选取不同的聚合物前驱体、模板和增强相,研发新型多尺度相改性碳泡沫复合材料,是实现材料多功能化的有效方法和途径。
本文以酚醛树脂为碳源,聚苯乙烯(PS)微球和酚醛微球为模板,纳米SiO2颗粒为增强相,通过模板法制备了纳米SiO2/碳微球多尺度相改性碳泡沫复合材料,研究了不同含量的SiO2颗粒对碳泡沫复合材料的力学性能、热性能的影响规律。
1 实 验
1.1 原料与试剂
苯酚(分析纯,天津市北联精细化学品开发有限公司);多聚甲醛(分析纯,天津市福晨化学试剂厂);氢氧化钡(分析纯,天津市天力化学试剂有限公司);十二烷基硫酸钠(SDS,分析纯,苏州腾豪化工科技有限那公司);苯乙烯(分析纯,济南铭信化工有限公司);过硫酸钾(KPS,分析纯,山东嘉源环保科技有限公司);SiO2颗粒(上海乃欧纳米科技有限公司);BJO-0930型酚醛微球(密度0.25 g·cm-3,平均粒径71.5 μm,平均壁厚1.84 μm,Asian Pacific Company[22])。
1.2 仪器
Quanta-450-FEG型扫描电子显微镜(英国牛津);X射线衍射仪(日本理学公司);Q500热重分析仪(美国TA公司);UTM5205X万能试验机(深圳三思纵横科技股份有限公司);TGL20M型高速离心机(湖南湘立科学仪器有限公司);TNX1200-30型马弗炉(上海向北实业有限公司);TL-1200型高温碳化炉(南京博蕴通仪器科技有限公司)。
1.3 材料制备
1.3.1 PS微球的合成
1)基于BIM的建筑工程设计管理可优化实践中的工程设计管理方式,使相关管理工作开展中实现对建筑工程信息数据的高效利用;
向三颈烧瓶中依次加入质量分数为23%的乙醇、72%的去离子水、4.75%的苯乙烯单体(St)和0.1%的乳化剂十二烷基硫酸钠,水浴加热至70 ℃,恒温5 min。再加入质量分数为0.15%的过硫酸钾,在一定的搅拌速率下,聚合反应10~12 h后,停止加热,反应结束。反应产物为乳白色液体,取其少量于离心管中,与适量的无水乙醇混合后反复离心、洗涤,于60 ℃下真空干燥24 h,即得到PS微球。
1.3.2 碳泡沫前驱体的制备
按1∶1.25物质的量之比,称取苯酚与多聚甲醛加入三颈烧瓶中,再加入质量分数为40%的Ba(OH)2溶液作催化剂,调节反应体系的pH值。此阶段先加入80%多聚甲醛;在70 ℃下反应60~70 min,再加入剩余多聚甲醛。随后升温至90~92 ℃,反应100~120 min,反应结束。待其冷却至室温,即得到热固性酚醛树脂。
以热固性酚醛树脂为基体,酚醛微球为分散相,PS微球与纳米SiO2颗粒为添加相,四者按一定比例混合均匀、注模、固化,即得到碳泡沫前驱体——酚醛泡沫。其中,酚醛微球、PS微球的质量分数分别为20%和10%,酚醛树脂质量分数为60%~70%,SiO2颗粒的质量分数分别为0%、5%和10%。
1.3.3 碳泡沫复合材料的制备
将上述前驱体置于高温碳化炉中,在N2保护氛围下,以1 ℃·min-1升温速率加热至850 ℃碳化-保温-缓慢冷却,即得到碳泡沫复合材料。为了对比SiO2颗粒含量对碳泡沫性能的影响,将前驱体中SiO2颗粒质量分数分别为0%、5%和10%的碳泡沫样品,依次记为Si-0、Si-1、Si-2。
1.4 表征与测试
1.4.1 微观结构
利用扫描电子显微镜对碳泡沫复合材料的微观结构及断口形貌进行表征,并用自带的X-MAX50型能谱仪进行微区元素分析;扫描电镜的工作电压为20 kV,工作范围为9~12 mm。采用日本理学X射线衍射仪分析多孔碳泡沫的相组成;40 kV管子电压,管流200 μA,扫描范围10~80°。
1.4.2 性能测试
采用万能试验机测试材料压缩性能,加载速率为0.25 mm·min-1,样品为直径10 mm的圆柱状。根据式(1)计算试样的压缩性能。
σ=F/S
(1)
式中:σ为压缩强度,MPa;F为试样承受的最大载荷,N;S为试样的横截面积,mm2。
采用热失重仪表征碳泡沫前驱体在N2氛围下的热失重行为,测试温度从室温到900 ℃,升温速率为10 ℃·min-1。并将样品热失重质量分数5%的温度,记录为起始分解温度[23]。
采用马弗炉对样品进行650 ℃下的等温氧化处理。用分析天平称量高温氧化前后样品的质量,由式(2)计算样品的氧化失重速率。每个样品测量3次,取其平均质量。
(2)
式中:R为氧化失重率;m1、m2分别为氧化前后试样的质量,精确到0.000 1 g。
2 结果与讨论
2.1 碳泡沫前驱体的热失重行为
图1为3种碳泡沫Si-0、Si-1、Si-2前驱体的热重曲线。
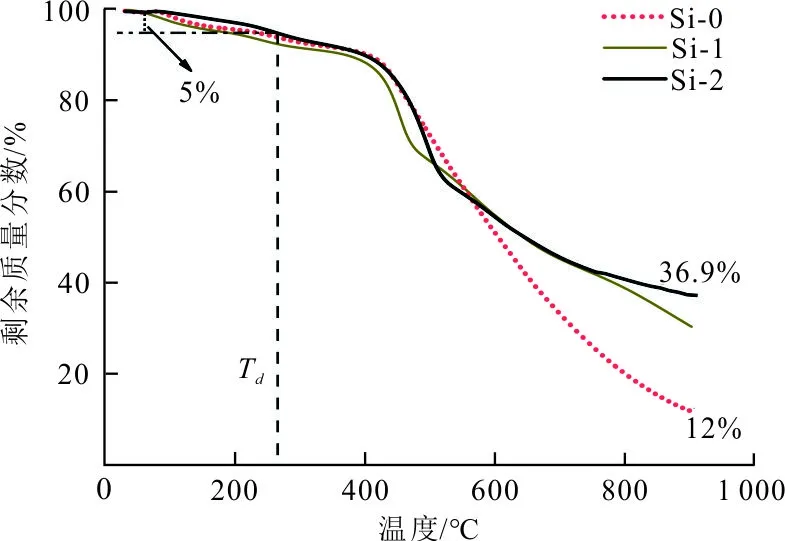
图1 碳泡沫前驱体的热失重曲线
可以看出,碳泡沫Si-2的前驱体的热稳定性最佳,其起始分解温度Td相对最高(265 ℃)。在900 ℃下的残碳率36.9%,比纯碳泡沫Si-0提高了207.5%。碳泡沫前驱体的热失重主要分为三阶段,以碳泡沫Si-2的前驱体为例,第一阶段为室温350 ℃,失重8.6%,这是由于在固化过程中,酚醛泡沫体残留的水分与低分子量混合物受热挥发所致,与此同时分子之间产生相互交联缩合[24-25],有少许失重。第二阶段为350~500 ℃,失重25.1%。这是由于大分子链断裂,使酚醛树脂发生裂解,释放出H2O、CO和CO2等挥发性物质。此阶段开始形成芳环结构[26],其中在453.6 ℃下的失重率尤为明显。第三阶段为500 ℃之后,失重28.3%,前驱体在此阶段不断发生结构重整、收缩。当温度超过800 ℃后,酚醛泡沫的失重趋于平缓,此时前驱体热解基本完成,挥发性物质大大减少,碳化反应基本结束,前驱体转变为无定形碳结构,碳泡沫形成。
2.2 碳泡沫复合材料的微观结构
图2为碳泡沫复合材料的SEM照片。可以看出,该复合材料以闭孔结构为主,主要由碳基体相、空心微球相、SiO2相和部分孔隙组成。其中,碳基体相彼此连接构成网状骨架结构;微球相较为均匀地分散在碳基体上,其主要为孔径为10~80 μm的闭孔球形结构,彼此几乎没有交联情况出现。孔隙主要分布在基体上,微球间也有少量的孔隙;孔隙的产生与前驱体系中的机械搅拌和热处理过程中的小分子的逃逸有关。

(a)Si-0 (b)Si-1
由图2(a)可看出,PS微球填充了原有的树脂与树脂基体相连的孔隙中,一定程度降低了孔隙率,从而提高了致密度。对样品Si-1中的点B进行EDS分析发现,该样品含有C、Si、O和少量Na元素。Si和O元素源于SiO2相,C和Na元素是制备PS微球过程中引入的SDS和KPS残留在碳泡沫中所致。另外,碳泡沫中存在一些微裂纹(见区域A、C),这主要是由于碳化过程中热应力未释放完全所致。
2.3 碳泡沫复合材料的压缩性能
图3为碳泡沫复合材料的压缩应力-应变曲线,可以看出不同含量的SiO2相对其压缩行为的影响。
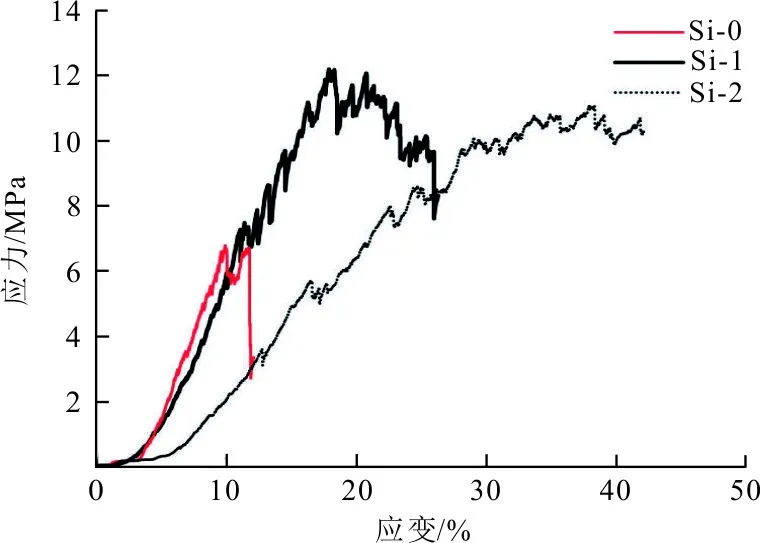
图3 碳泡沫复合材料的压缩应力-应变曲线图
从图3可看出,不含纳米SiO2相的碳泡沫Si-0,表现出一定的脆性断裂特征,应变为12%处出现显著的应力骤降(衰减61.5%)。当引入少量纳米SiO2相,碳泡沫Si-1屈服阶段的应力衰减现象有所减缓,其断裂模式逐渐变为梯度脆性断裂,可以看到,碳泡沫复合材料的压缩过程要分为弹性形变区和屈服平台区;在最初受力时呈现弹性形变,随后进入平台区。其压缩状态下的应力张量,可由垂直于载荷方向的静应力和载荷方向45°的剪应力所组成[27]。初遇载荷时,应力线性增大,压应力使微裂纹在碳泡沫空隙中不断扩展;当扩展的裂纹遇到纳米SiO2颗粒时发生裂纹偏转,这可使碳泡沫承受更大的应力,同时消耗一定的微裂纹扩展能量,在应力-应变曲线中表现为屈服平台区的“锯齿状”[28]。
当纳米SiO2相含量进一步增大,碳泡沫Si-2的应力-应变曲线无明显的应力衰减现象,虽压缩强度有所下降,但断裂韧性提高较为明显,其断裂特征向假塑性断裂方式转变。可见,适量的纳米SiO2颗粒有助于材料的强度的提高,并在一定程度上改善了材料的塑韧性。
材料的强度受与密度有关,为了消除其密度对强度的影响,计算碳泡沫复合材料的比压缩强度,结果见表1。

表 1 碳泡沫复合材料的压缩强度与比压缩强度
随着SiO2颗粒含量的增加,碳泡沫的压缩强度呈先升高后略降低的趋势。当纳米SiO2质量分数为5%时,其压缩强度最大,为12.2 MPa。纳米SiO2含量对碳泡沫比压缩强度的影响规律与压缩强度相似,亦为碳泡沫Si-1最大,该样品的比压缩强度为33.5 MPa·cm3·g-1,比不含纳米SiO2的碳泡沫Si-0提高了65.3%。
图4为碳泡沫Si-1的压缩断口形貌。为清晰地解释纳米SiO2粒子对断口形貌的影响,图4(b)选择背散射(BSE)照片。可以看到,碳泡沫的压缩断口并不平整。在受压过程中碳泡沫内部出现了若干较长的裂纹,当断裂端受到的横向拉应力与结合强度相等,裂纹突然扩张,导致微球相周围的应力重新分布,进而加速裂纹扩展[29-30]。将区域D放大,可清晰看到裂纹扩展方向并不沿同一平面,裂纹扩展过程中遇到微球相、团聚的SiO2相(区域E所示)和分散SiO2(红色箭头位置),发生一定的偏转,裂纹尖端应力弱化,碳泡沫得以继续承受载荷,完成平台区的压缩变形过程,体现为图3应力-应变曲线中的“锯齿状”,表现为梯度脆性断裂的特征。
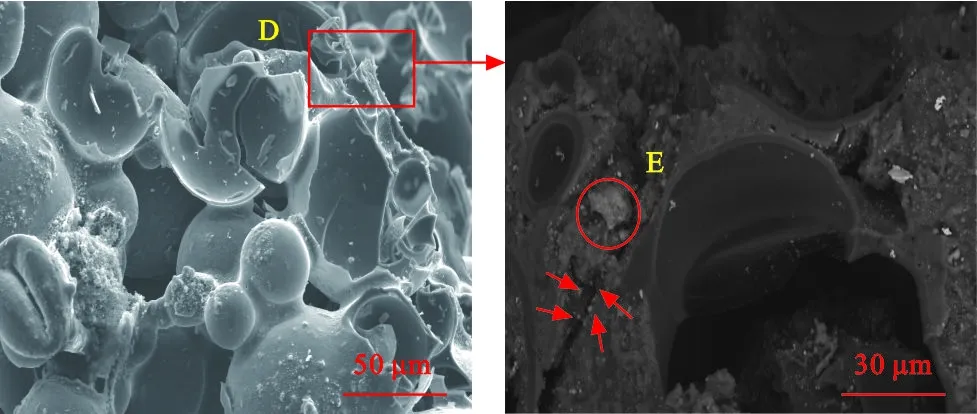
(a)Si-1 (b)区域D的断口放大图
2.4 碳泡沫复合材料的耐高温氧化性能
对碳泡沫复合材料进行650 ℃等温氧化25 min。不同含量纳米SiO2颗粒对碳泡沫复合材料热氧化性能的影响,如图5所示。
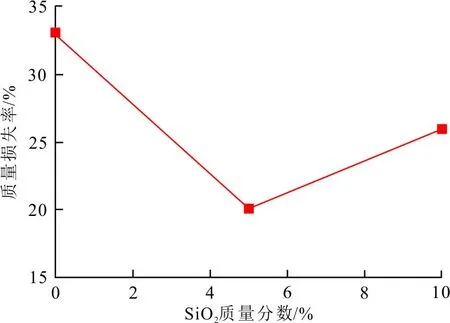
图5 SiO2含量对碳泡沫耐热氧化性能的影响
可以看出,引入SiO2相,对碳泡沫的抗氧化能力有所提高,其质量损失率显著降低。其中,碳泡沫Si-1的耐高温氧化性能最佳。与纯碳泡沫相比,其质量损失由33%降低为20%,耐高温抗氧化能力提高了39.4%。这是由于部分SiO2相分布在碳基体孔隙中,见图2(b)和图4(b),其热导率较低,阻挡了热量传递及热氧的渗透。但过多的纳米SiO2相易发生团聚,造成微球相和碳基体相的裸露。当SiO2相进一步提高,碳泡沫复合材料的抗氧化能力有所下降,碳泡沫Si-2的耐高温氧化能力较纯碳泡沫Si-0仅提高了10.4%。
图6为碳泡沫Si-1热氧化25 min后的SEM照片。可以看到,部分微球相被氧化,出现明显的空心状,材料内部亦出现了明显的孔洞。但由于SiO2相起到一定的抗氧化作用,碳泡沫总体保持相对完整的形貌。

图6 Si-1热氧化后的SEM照片
3 结 论
本文以PS球、空心微球为模板,树脂基体为碳源,SiO2相为增强相制备了碳泡沫复合材料。其中,闭孔微球相均匀分布在树脂碳基体上,SiO2相分布在碳基体的孔隙中。SiO2相的引入,使得碳泡沫复合材料在承受应力时,消耗了一定的微裂纹扩展功,阻碍裂纹进一步扩展,提高了碳泡沫的强度,同时提高了其耐高温热氧化性。当SiO2质量分数为5%时,碳泡沫的压缩强度和比压缩强度均达到最大,为12.2 MPa和33.5 MPa·cm3·g-1,较改性前分别提高76.8%和65.3%。同时,该样品的耐高温氧化性能相对最优。与纯碳泡沫相比,其在650 ℃下氧化25 min的耐高温抗氧化能力提高了39.4%。
猜你喜欢
环球时报(2023-03-22)2023-03-22 15:18:28
作文周刊·小学一年级版(2022年20期)2022-05-07 01:15:17
趣味(数学)(2021年4期)2021-08-05 07:58:46
潍坊学院学报(2020年6期)2020-11-22 08:04:10
材料科学与工程学报(2016年1期)2017-01-15 13:33:52
当代化工研究(2016年7期)2016-03-20 16:21:54
大连工业大学学报(2015年4期)2015-12-11 04:06:50
电源技术(2015年9期)2015-06-05 09:36:06
中国当代医药(2015年29期)2015-03-01 02:07:41
创业家(2015年9期)2015-02-27 07:54:39
