
幼年型粒单核细胞白血病患者5例临床及基因分析
2022-02-16 07:33陆海燕王晓欢程艳丽索涛莉山西省儿童医院血液科太原030001通讯作者mail2902196190qqcom
山西医科大学学报 2022年1期
陆海燕,王晓欢,程艳丽,王 静,索涛莉(山西省儿童医院血液科,太原 030001;通讯作者,E-mail:2902196190@qq.com)
幼年型粒单核细胞白血病(JMML)起源于造血干细胞,兼有骨髓增生异常综合征及骨髓增殖性疾病特征,临床表现及预后有很大的异质性[1]。JMML是一种罕见的发生于儿童的慢性髓系白血病,发病率仅为1.2/106,临床接诊此类患儿很少[2]。现将我科近3年诊治的5例JMML资料总结如下。
1 资料及方法
1.1 研究对象
收集2018年5月至2021年7月在山西省儿童医院血液科诊断为JMML患儿5例,均符合2016年WHO制定的JMML诊断标准[3]。
1.2 方法
临床诊断JMML患儿,采集骨髓标本2 ml(EDTA抗凝),提取DNA,用Life Technologies平台S5半导体测序仪进行检测,所得原始数据选择Torrent Server中Coverage Analysis插件进行覆盖度分析,原始数据上传Ion Reporter服务器,人工检索与筛选,报告蛋白酪氨酸磷酸酶非受体型11(protein tyrosine phosphatase non-receptor type 11,PTPN11)基因、卡西塔斯B系淋巴瘤(Casitas B-lineage lymphoma,CBL)基因、神经纤维肉瘤Ⅰ型(neurofibromatosis 1,NF1)基因、大鼠肉瘤病毒(rat sarcoma,RAS,包括NRAS、KRAS)基因突变(由第三方实验室检测)。收集患儿诊断时年龄、发病到诊断时间、主要临床表现、血系列、外周血涂片、胎儿血红蛋白、骨髓形态、BCR/ABL基因及染色体,定期随访治疗及预后。本基因检测获得患儿监护人知情同意。
2 结果
2.1 一般情况及临床表现
诊断时中位年龄23月,发病到诊断中位时间2月,最后随访时间为2021年8月15日,中位随访时间6月。临床表现:5例脾大(2例脾大入盆);5例肝大,多为轻中度增大;4例有斑丘疹样皮疹;腹胀3例;皮肤出血点及面色苍白各2例;发热、反复口腔溃疡及下肢跛行各1例(见表1)。
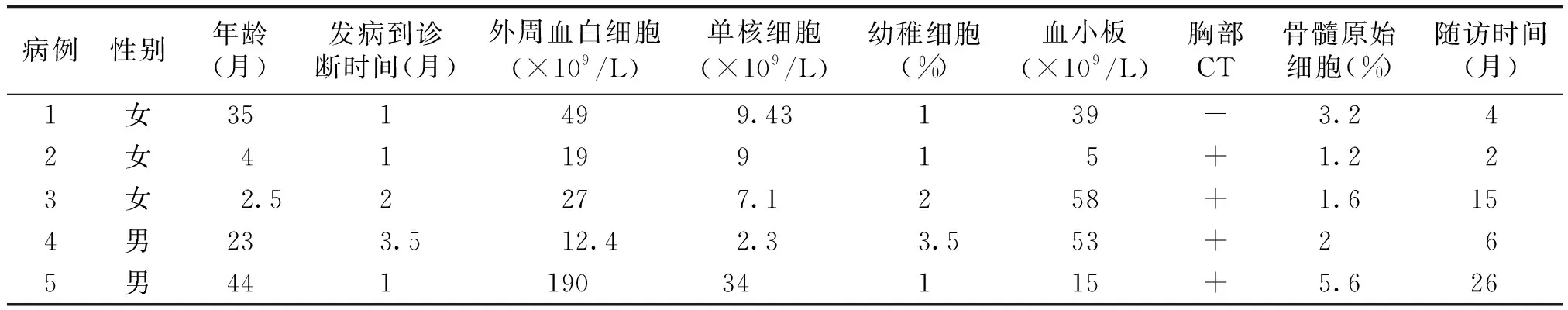
表1 JMML患儿5例的临床资料
2.2 辅助检查
外周血白细胞中位计数27×109/L,单核细胞中位计数9×109/L,外周血幼稚细胞中位计数2.5%。血红蛋白F(HbF)高于同年龄正常值4例。骨髓形态明显或极度增生活跃,粒系占59.6%~84.8%,均有轻度病态造血,原始粒细胞比例1.2%~5.6%。染色体4例正常,1例为46,XX,inv(9)(p12,q13),为原发性异常(见表1)。
2.3 基因检测结果
3例检测到PTPN11基因突变,突变位点为Exon3、Exon13;2例检测到KRAS基因突变,突变部位为Exon2;5例均为体细胞杂合错义突变(见表2,图1,图2)。
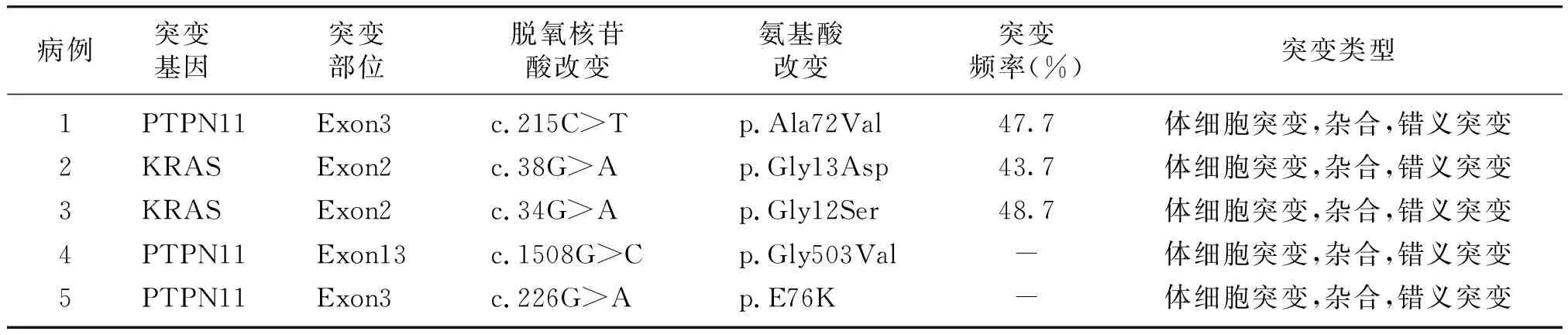
表2 JMML患儿5例基因检测结果
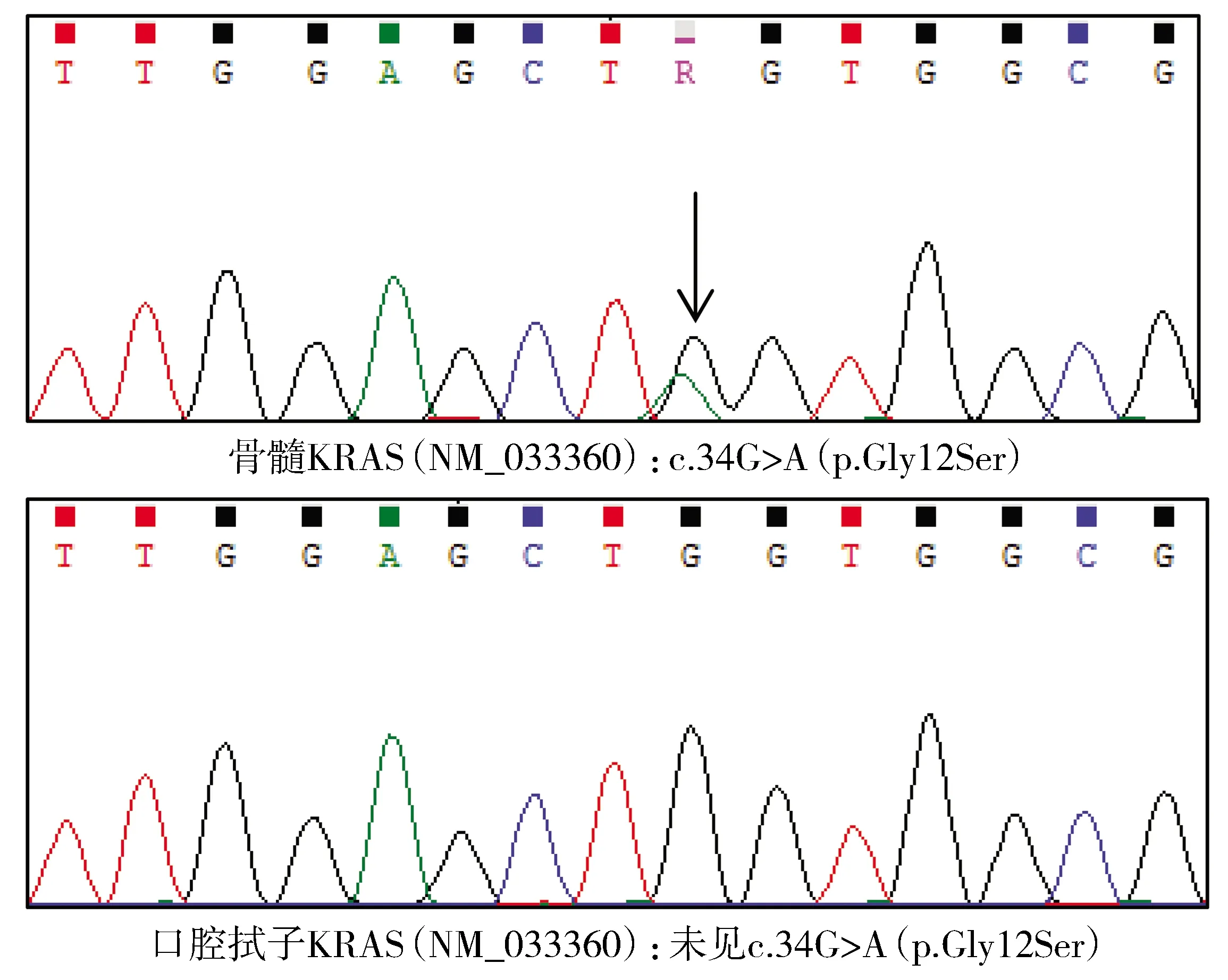
图1 1例JMML患儿检测到KRAS体细胞突变Figure 1 KRAS somatic mutation of one child diagnosed with JMML
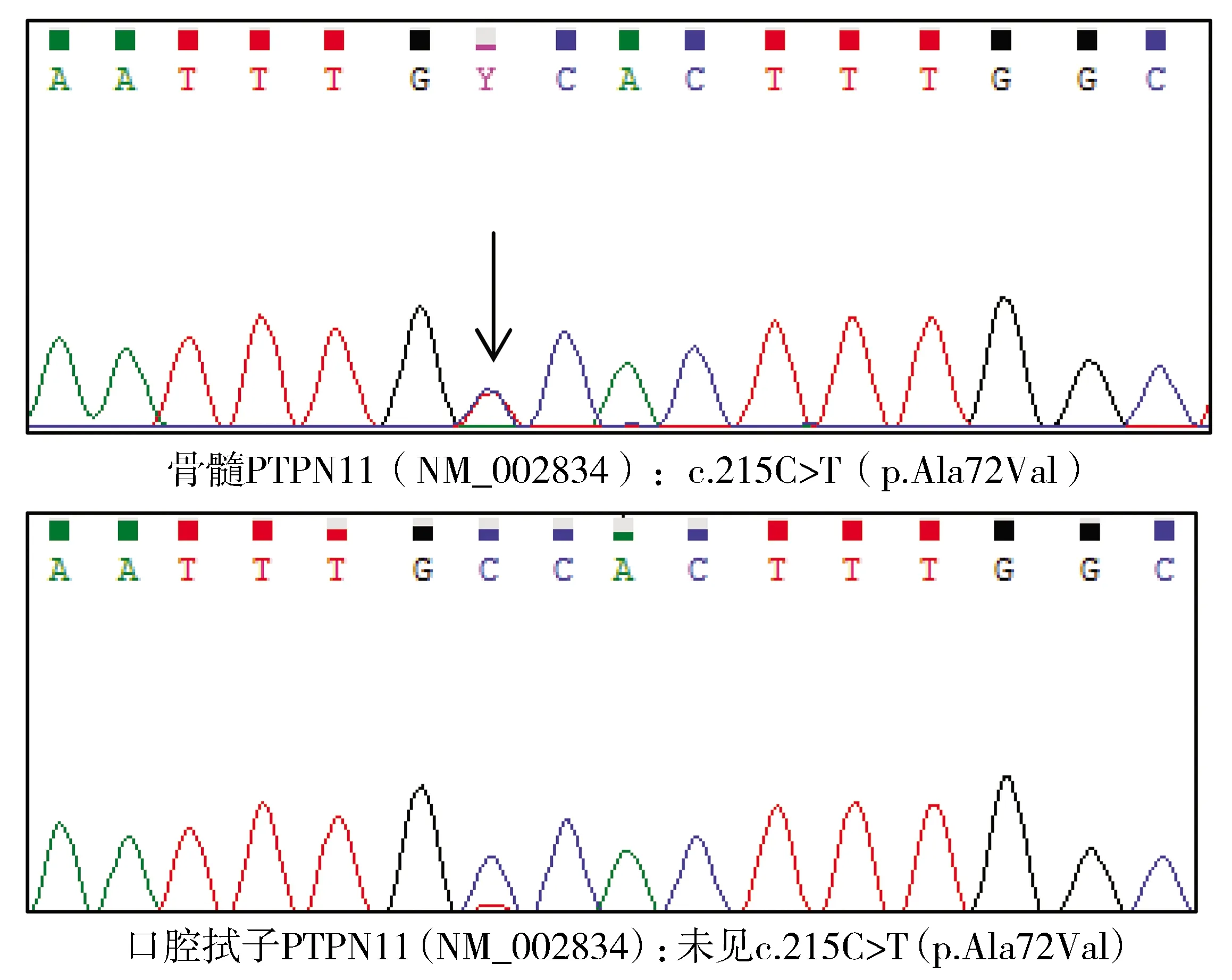
图2 1例JMML患儿检测到PTPN11体细胞突变Figure 2 PTPN11 somatic mutation of one child diagnosed with JMML
2.4 治疗及转归
1例PTPN11突变患儿口服中药及支持治疗,随访6月,病情进展合并颅内出血死亡;1例PTPN11突变患儿口服乐疾宁治疗,外周血细胞一度改善,肝脾缩小,治疗4月合并肺炎、肺出血,呼吸衰竭死亡;1例PTPN11突变患儿,诊断后口服西罗莫司治疗,后行父供子单倍体移植(外院),目前移植后2年,原发病持续缓解。2例KRAS突变均为小婴儿,确诊后未治疗,1例合并CMV感染行更昔洛韦抗病毒治疗,外周血白细胞、单核细胞下降,血小板上升,肝脾明显回缩,截止目前共随访15月;另1例患儿诊断时血小板<1×109/L,因血小板输注无效,放弃治疗后2月合并颅内出血死亡。
3 讨论
JMML是一种侵袭性克隆性造血异常,以粒单核系过度增殖为特征。多发生于婴儿及儿童早期,中位诊断年龄为2岁,大部分在4月至4岁确诊,男女比例为2 ∶1-3 ∶1[1]。JMML临床表现多由粒单系细胞浸润引起,常见受累脏器为肝脾、皮肤、肺部、胃肠道等。
JMML发病主要与多种突变基因引起RAS/促细胞分裂原活化蛋白激酶(MAPK)信号通路异常有关。RAS下游通路Raf/MEK/ERK、PI3K/Akt/mTOR及RalGDS的激活参与疾病形成。90%病例存在RAS基因(KRAS及NRAS)及调控基因(PTPN11、CBL及NF1)突变,PTPN11基因体细胞突变见于35% JMML病人[1,3,5]。Liao等[4]发现NF1基因突变最常见,其次为NRAS、PTPN11及CBL基因突变。PTPN11基因突变多为外显子3,4,13的点突变或错义突变,E76K是JMML中最常见的体细胞PTPN11突变类型[1,5]。本研究中3例PTPN11基因突变2例发生于3号外显子,1例发生于13号外显子,其余2例KRAS基因突变发生于2号外显子。表观遗传学异常也参与JMML发生,JMML特定DNA区域,尤其抑癌基因启动子区域CpG岛有高甲基化[2]。2011年Olk-Batz等[6]对127例JMML患儿甲基化水平进行测量,发现了4个高频甲基化的基因(BMP4基因、CALCA基因、CDKN2B基因及RARB基因)。多基因高甲基化与SET结合蛋白1(SETBP1)基因和Janus激酶3(JAK3)基因的继发突变有显著关联。RAS通路信号激活与甲基化之间可能存在功能联系[7]。
诊断时年龄>2岁、HbF>10%、血小板<33×109/L是目前公认的JMML预后的危险因素。基因突变类型与预后有一定相关性,PTPN11突变预后差,CBL胚系突变常会发生自发缓解,KRAS突变及大部分NRAS突变病情会进行性恶化[8,9]。单纯化疗对JMML疗效甚微,HSCT前行化疗可减轻疾病负荷,HSCT前后使用去甲基化药物可改善病情、预防复发,但目前尚无定论[10-13]。靶向抑制RAS/MAPK通路相关蛋白的药物,多在临床试验中[14]。造血干细胞移植(HSCT)是目前唯一可能根治JMML的措施。我们统计的5例患儿中4例存在高危因素,中位随访6月,除1例行HSCT后病情控制,其余均死亡或进展。少数RAS突变会自发缓解,赵卫红报道1例NRAS体细胞突变的JMML患儿未治疗存活10年[15]。我们1例2.5月患儿,HbF为年龄范围正常值,血小板>33×109/L,为KRAS体细胞突变,对症治疗后病情改善,随访15月,由于时间短是否会自发缓解还需进一步随访。
JMML多发生于婴幼儿,多有RAS基因及其调控基因突变,不规律治疗多预后差,KRAS体细胞突变且不合并高危因素婴儿有自发缓解可能。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
中国生殖健康(2020年4期)2021-01-18
新农业(2020年22期)2020-12-18
今日农业(2020年24期)2020-12-15
中外文摘(2020年13期)2020-08-01
福建基础教育研究(2020年4期)2020-05-28
时代英语·高三(2019年4期)2019-09-03
肿瘤预防与治疗(2019年6期)2019-07-30
科学中国人(2015年23期)2015-07-12
中学生理科应试(2014年12期)2015-01-15
