
肌力下降、呼吸衰竭、心力衰竭、抗线粒体抗体阳性
——罕见炎性肌病的不典型表现
2022-02-16 02:31孙晓红郭潇潇留永健彭琳一王怡宁曹宇泽刘跃华赵肖奕
协和医学杂志 2022年1期
路 菲,张 宁,康 琳,孙晓红,田 然,郭潇潇,留永健,张 文,彭琳一,苏 童,曹 剑,王怡宁,钱 敏,曹宇泽,杨 璐,刘跃华,张 路,赵肖奕,冷 泠
中国医学科学院北京协和医院 1老年医学科 2心内科 3呼吸与危重症医学科 4风湿免疫科 5放射科6神经科 7皮肤科 8康复医学科 9医学科学研究中心干细胞生物学平台,北京 100730
1 病历简介
患者女性,65岁,主诉“双下肢无力8年,喘憋、下肢水肿4个月”,于2020年11月5日收住北京协和医院老年医学科。
1.1 现病史
8年前患者无明显诱因出现双下肢无力,表现为抬腿、骑车困难,上楼费力,未诊治。半年前出现进食有哽噎感,未诊治。患者2020年6月受凉后出现咳嗽、咳白色泡沫痰,无发热。自服“清开灵”“阿莫西林”3 d后症状减轻。此后平地行走(约100 m)出现喘憋,休息10~20 min后症状逐渐缓解;夜间入睡不能平卧,偶有睡眠中憋醒。上述症状间断发作,约2~3次/月,并逐渐出现双足-足踝-小腿对称凹陷性水肿,且进行性加重。2020年7—8月,患者先后至当地医院住院治疗2次。首次入院血常规示白细胞(white blood cell, WBC)7.45×109/L,中性粒细胞(neutrophil, NEUT)百分比65.4%,血红蛋白(haemoglobin,HGB)133 g/L,血小板(platelet, PLT)300×109/L;血生化示谷丙转氨酶(alanine aminotrans-ferase, ALT)41.9 U/L↑,γ-谷氨酰转肽酶(gamma-glutamyl transpeptidase, GGT)362 U/L↑,碱性磷酸酶(alkaline phsphatase, ALP)312 U/L↑,超敏C反应蛋白3.0 mg/L,氨基末端脑钠肽前体(N terminal-pro B type natriuretic peptide, NT-proBNP) 1180 ng/L↑。胸部CT平扫:双肺炎症、纵隔淋巴结肿大,右肺上叶磨玻璃结节、中叶膨胀不全,左肺上叶局限性肺气肿。超声心动图:左心房增大,钙化性瓣膜病,三尖瓣反流(轻度),左心室充盈功能减低,左室射血分数(left ventricular ejection fractions, LVEF)60%。动态心电图:窦性心动过速,Ⅰ度房室传导阻滞(atrioventricular block,AVB),偶发房性早搏,频发室性早搏(可见成对或多源)。诊断为“肺部感染,心力衰竭”。予以抗感染、利尿、纠正心力衰竭等治疗(具体药物不详)后症状无明显减轻,仍有喘憋(步行<10 m即出现)。为进一步诊治收入北京协和医院老年医学科病房。
患者起病以来,精神弱、睡眠差,饮食可,大便正常,尿量无明显减少。体力进行性下降,体质量无明显增减。
1.2 既往史、个人史及家族史
患者20年前无明显诱因出现双眶周皮肤色素沉着、双侧眉周毳毛增多,并逐渐加重,未系统诊治。高血压15年,最高214/100 mm Hg(1 mm Hg=0.133 kPa)。长期服用缬沙坦(80 mg×1次/d)、倍他乐克(25 mg×1次/d),平素血压控制于(130~160)/(80~100) mm Hg。2型糖尿病10年,长期服用二甲双胍(0.5 g×2次/d)、门冬胰岛素30注射液(早14 IU、晚10 IU)皮下注射,自测空腹血糖5.0~9.0 mmol/L,餐后血糖13.0~18.0 mmol/L。血脂升高8年,长期服用辛伐他汀(10 mg×1次/d)降脂治疗,3年前改为瑞舒伐他汀(10 mg×1次/d),未规律监测血脂水平。2012年11月跌倒致腰椎骨裂,保守治疗后好转。个人史、婚育史无特殊。家族史:父亲因“肝癌”去世,母亲因“心脏病”去世;有1姐2兄,均为2型糖尿病患者。
1.3 入院查体
体温36.3 ℃,呼吸30次/min,心率95次/min,血压140/81 mm Hg,血氧饱和度为89%~91%。发育正常,营养中等,神志清楚,端坐位,呼吸短促。眶周皮肤色素沉着、眉周毳毛增多(图1)。双侧颈静脉怒张。双侧胸廓运动度减低,胸椎后凸,站立时骨盆后倾。双下肺闻及呼气末少量细湿啰音及爆裂音。心律不齐,每分钟闻及5~6次早搏,各瓣膜听诊区未闻及杂音。腹壁质韧,全腹无压痛、反跳痛、肌紧张,听诊肠鸣音3~4次/min。双下肢对称性凹陷性水肿,双手细颤,食指为著。四肢肌力Ⅴ-,肌张力正常,双侧病理征(-)。
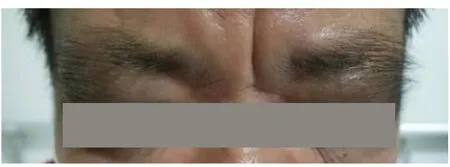
图 1 患者双侧眶周皮肤色素沉着伴眉周毳毛增多
1.4 实验室检查
血常规:WBC 9.94×109/L↑,NEUT百分比65.7%,HGB 132 g/L,PLT 370×109/L。
尿常规+尿沉渣检查:WBC 71.8/μL↑,BACT 603.1/μL↑,红细胞 1.5/μL,尿蛋白1.0 g/L;24 h尿蛋白定量0.92 g↑。便常规+潜血(-)。
血生化:ALT 64 U/L↑,谷草转氨酶(aspartate aminotransferase, AST) 53 U/L↑,GGT 337 U/L↑,ALP 495 U/L↑,血磷 1.62 mmol/L↑,白蛋白35 g/L,前白蛋白 119 mg/L↓。凝血功能:纤维蛋白原4.28 g/L↑,D-二聚体0.74 mg/L↑。
动脉血气分析:pH值7.38,PaCO263 mm Hg↑,PaO238 mm Hg↓,HCO3-34.2 mmol/L↑,剩余碱8.7 mmol/L↑,乳酸 1.1 mmol/L。
心肌损伤标志物及心肌酶:心肌肌钙蛋白Ⅰ 0.062 μg/L↑,脑钠肽 289 ng/L↑,NT-proBNP 1241 ng/L↑,肌酸激酶(creatine kinase, CK)116 U/L,乳酸脱氢酶 150 U/L,肌红蛋白 92 μg/L。
肿瘤标志物:甲胎蛋白 25.2 μg/L↑,糖链抗原125 38.8 kU/L↑,余无异常。
感染四项及炎症指标:均未见异常。
免疫相关指标:血补体+免疫球蛋白示IgM 2.64 g/L↑,补体(-)。血清蛋白电泳示α2 11.2%↑;血轻链 KAP 1420 mg/dL↑,LAM 699 mg/dL↑,比值2.03。尿轻链 KAP 17.40 mg/dL↑,LAM 6.73 mg/dL↑。血清免疫固定电泳:κ 31.4 mg/L↑,λ 51.5 mg/L↑,κ/λ 0.610,尿免疫固定电泳(-)。抗核抗体17项示抗核抗体(antinuclear antibody, ANA)(+)胞浆型1∶320,抗ds-DNA抗体IgG(+)150,抗Ro52(+++)。自身免疫性肝炎抗体示ANA(+)胞浆型1∶320,抗线粒体抗体(anti-mitochondrial antibody, AMA)(+)1∶320,AMA-M2(+)>400。抗中性粒细胞胞质抗体(-)。肌炎抗体谱(-)。抗线粒体亚型抗体示AMA-M2弱(+),M4、M9亚型均(-)。
内分泌相关指标:甲状腺功能指标正常;血清总皮质醇38.1 μg/dL↑,24 h尿游离皮质醇、血促肾上腺皮质激素、α-葡萄醛酸酶活性均正常。
1.5 辅助检查
胸部CT平扫:右侧膈肌显著抬高,右肺中叶膨胀不全,双下肺透亮度降低,伴间质纹理增多,左心房显著扩大,食管扩张、内有液体。腹部及盆腔CT平扫未见明显异常。
肺功能(通气):限制性通气功能障碍。
心电图:窦性心律,心率86次/min,可见室性期前收缩。动态心电图:总心搏数131 407次,房性早搏<0.1%,可见房性早搏未下传,室性早搏15 403(11.72%),可见成对室性早搏349对、室性心动过速210阵、二联律504阵、三联律95阵,Ⅰ度AVB。
超声心动图:室间隔基部及中段运动稍减低,左心房增大(前后径 49 mm),升主动脉增宽(近端升主动脉内径 37 mm),主动脉瓣及二尖瓣后叶瓣环退行性变,轻度主动脉瓣及二尖瓣关闭不全,轻度肺高血压(估测肺动脉收缩压 42 mm Hg)。
冠状动脉CT血管造影:冠状动脉呈右优势型、重度钙化;左主干未见明确狭窄;前降支、右冠状动脉多发混合斑块,管腔轻-中度狭窄;回旋支近中段混合斑块,管腔轻度狭窄。心肌核素显相(静态)未见明显异常。
肌电图:上下肢可见自发电位,多相波比例增高,重复神经电刺激未见异常。
膈肌超声:双侧膈肌移动幅度减低,右侧为著(左侧2.3 cm、右侧1.8 cm)。
MRI:大脑左侧基底节区腔隙灶;双侧放射冠、半卵圆中心多发斑片状异常信号,慢性缺血性改变;垂体饱满。T2WI回旋序列示心肌信号未见明显异常,左右心房增大,室间隔略增厚,室间隔、左室壁运动减低。T1和T2 mapping序列示T1、T2信号均增高(T1波动于1348~1699 ms,T2波动于38~52 ms,图2),以T1升高为主,考虑心肌组织纤维化可能性大;T2轻度升高,考虑伴有轻度水肿改变。双侧大腿各组肌肉及皮下脂肪T2压脂序列高信号,考虑炎性病变可能(图3),双侧大腿肌肉容积减少,尤其大腿后部肌群见斑驳状脂肪浸润,提示肌肉萎缩。

图 2 心脏MRI mapping序列示心肌T1和T2信号绝对值均增高
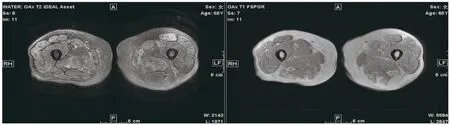
图 3 MRI示双侧大腿各组肌群及皮下脂肪在T2压脂像均呈现弥漫性异常高信号
股四头肌活检HE染色示横纹肌组织轻度变性;刚果红(-),高锰酸钾化刚果红(-),醇化刚果红(-)。腹壁皮肤活检:真皮及皮下组织小血管管壁增厚,真皮全层小血管周围未见红染物质沉积,真皮深部汗腺导管及腺腔部分红染,皮下脂肪组织小血管管壁极少量散在阳性,考虑为非特异性染色。
2 第一次多学科讨论(2020- 11- 25)
2.1 老年医学科
患者为老年女性,慢性病程,因“心功能不全”入院。肺部CT示右侧膈肌明显抬高,食管扩张。患者于2012年无明显诱因出现双下肢无力,进行性加重,2020年3月起伴进食哽噎感,偶有饮水呛咳。结合辅助检查结果,患者存在多系统受累:(1)心脏:入院时有喘憋、不能平卧、双下肢水肿,血脑钠肽、NT-proBNP升高,为心力衰竭的表现;同时合并心律失常,主要为频发室性早搏,伴短阵室性心动过速,Ⅰ度AVB。(2)呼吸系统:双肺炎症性改变,突出表现为Ⅱ型呼吸衰竭、二氧化碳潴留。但患者既往无慢性阻塞性肺疾病、阻塞型睡眠呼吸暂停综合征等呼吸系统病史,其二氧化碳潴留难以用肺部疾病解释。(3)肌肉系统:患者病程中出现进行性双下肢无力,膈肌超声示双侧膈肌运动幅度减低;MRI见双侧大腿各组肌群T2压脂像弥漫性异常高信号及肌肉容积减少。同时近半年出现吞咽哽噎感,胸部CT见食管扩张,提示存在骨骼肌受累及可疑食管平滑肌受累。(4)自身免疫系统:存在抗核抗体、AMA、AMA-M2、及抗Ro-52抗体高滴度阳性,伴胆管酶水平升高,且胆管酶升高程度与其他肝酶水平不匹配。(5)皮肤及毛发异常:最突出的面部特征为双侧眶周色素沉着、眉周毳毛增多,上述改变在患者中年后逐渐出现。多学科讨论需解决的问题:(1)多系统受累可否用一元论解释?(2)下一步治疗方案?
2.2 心内科
患者具有全心衰竭表现且心肌损伤标志物升高,考虑存在心肌损伤。心脏节律方面,存在Ⅰ度AVB,多源室性早搏。心脏MRI及超声心动图示心肌纤维化、左右心房增大、轻度肺动脉高压。冠状动脉CT血管造影示多发粥样硬化斑块伴狭窄,但血流TIMI为Ⅲ级,心电图未见陈旧心肌梗死表现,静态心肌核素现象未见心肌灌注异常。目前考虑诊断:(1)稳定性冠心病、双支病变(累及前降支、回旋支);轻度肺动脉高压;心律失常、房性早搏、多源性室早。(2)系统性疾病心脏受累:心肌、骨骼肌损伤明确,目前无明确淀粉样变证据。结合心脏MRI,考虑炎性肌病的可能。建议行心肌活检及右心漂浮导管检查明确肺动脉高压病因,以利于进一步诊断。治疗方面,建议予以利尿以减轻容量负荷,小剂量β受体阻滞剂以减少室性早搏。
2.3 呼吸与危重症医学科
呼吸系统主要表现为Ⅱ型呼吸衰竭。患者平素无喘鸣,肺部听诊无干啰音,肺功能检查未见气流阻塞,第1秒用力呼气容积(forced expiratory volume in one second, FEV1)为88%,但存在限制性通气功能障碍,故考虑是由于呼吸空间受限或呼吸肌力量不足导致的肺泡低通气,其常由肺外疾病所致:(1)胸膜腔空间被挤占:如胸腔积液、气胸、腹部疾病(腹腔积液等)导致膈肌显著升高。(2)胸壁结构异常:如胸廓畸形、脊柱侧凸、重度肥胖等。(3)神经肌肉疾病:膈肌麻痹或全身病变累及呼吸肌。患者胸部CT主要异常为右侧膈肌明显抬高,提示可能存在膈肌功能障碍;另可见食管扩张、有较多液体残留,提示食管平滑肌亦可能受累。患者近年有逐渐加重的肢体无力,平卧位呼吸困难(表现为显著的吸气无力)。CT特征与临床表现相符,超声进一步证实了膈肌活动度明显下降。综上,考虑全身性神经肌肉疾病,呼吸肌是受累部位之一。治疗方面,建议在神经肌肉疾病改善前,长期应用双水平气道正压通气(bilevel positive airway pressure,BiPAP)呼吸机改善肺通气。目前经BiPAP治疗,患者呼吸困难症状已显著改善。复查动脉血气分析虽仍存在二氧化碳潴留,但pH值已恢复正常。对于该患者,不能追求完全正常的二氧化碳分压,而需关注其pH值是否可维持在7.35以上。建议BiPAP夜间持续使用,白天视呼吸困难程度酌情使用。
2.4 神经科
肌力下降以肢体近端为著,膈肌运动幅度明显减低,肌电图示上下肢自发电位,多相波比例增高,结合MRI表现考虑定位诊断在肌肉。至于定性诊断,由于存在膈肌麻痹,可见于以下疾病:(1)脊髓疾病:如颈椎破坏性病变;(2)运动神经元病:如肌萎缩侧索硬化;(3)周围神经病:如吉兰-巴雷综合征(Guillain-Barrés syndrome, GBS)、病毒感染后神经病变;(4)神经肌肉接头疾病:如重症肌无力;(5)肌肉疾病:如多肌炎、皮肌炎、肌营养不良、代谢性肌病;(6)全身性疾病:甲状腺功能减退/亢进、结缔组织病引起的肺减缩综合征[1]等。结合患者合并心脏受累,考虑肌营养不良、先天性肌病、代谢性肌病(糖原累积病、脂类代谢肌病、线粒体肌病等)、全身性疾病(副肿瘤综合征、自身免疫性疾病、淀粉样变等)。老年患者,病程较长,症状相对轻,而肌营养不良通常有特定的累及部位,故可能性较小。先天性肌病多幼年起病,且症状突出,亦可排除。综合目前检查结果,副肿瘤综合征、内分泌疾病、淀粉样变、病毒感染的证据均不充分,应重点考虑为代谢性肌病(患者α-葡萄糖苷酶结果为阴性,可排除糖原累积病)。该类肌病可发生于各个年龄段,具有运动不耐受的表现。患者股四头肌活检HE染色无特异性提示,可进一步送检神经病理实验室进行特殊染色。如有异常,可进一步完善基因检测明确疾病类型。此外,需排除全身性疾病肌肉受累的情况。
2.5 风湿免疫科
老年女性,存在心肌、呼吸肌、吞咽肌及骨骼肌受累,目前感染、肿瘤、内分泌、电解质紊乱相关肌病的证据不足,其原发病考虑为特发性炎性肌病(idopathic inflammatory myopathy, IIM)、肌肉疾病(肌营养不良)及药物相关肌病。患者病程持久,药物相关肌病可能性小。根据1975年 Bohan和Peter 分类标准[2]和2017年欧洲抗风湿病联盟/美国风湿病学会分类标准[3],确诊多发性肌炎(polymyositis,PM)需满足:(1)肢带肌(肩胛带、骨盆带、四肢近端肌)和颈前屈肌对称性无力,可伴有吞咽困难和呼吸肌无力;(2)典型皮疹;(3)血清肌酶升高;(4)肌电图呈肌源性改变;(5)肌活检有特征性异常表现,且无其他肌病组织病理学表现。目前该患者仅符合第一条诊断标准,不具有炎性肌病的典型表现。但炎性肌病可累及上述肌群,且患者存在抗Ro52阳性、AMA-M2阳性,炎性肌病仍不能排除。为明确诊断,建议股四头肌活检标本送检神经病理实验室进行特殊染色。若条件允许,可完善正电子发射断层显像/计算机体层成像(positronemission tomogra-phy /computed tomography, PET/CT)检查评估有无肿瘤、肌肉炎症的证据。治疗方面,若排除淀粉样变、肿瘤,可尝试免疫抑制治疗,给予糖皮质激素和/或人免疫球蛋白治疗。但该患者合并冠心病、高血压、糖尿病等基础疾病,应充分权衡激素治疗的利弊。此外,患者存在以胆管酶升高为主的肝功能异常,血清IgM轻度升高,AMA-M2阳性,肝脏超声未发现肝外梗阻,无病毒性肝炎证据,符合2009年原发性胆汁性胆管炎(primary biliary cholangitis, PBC)诊断标准[4],可给予熊去氧胆酸胶囊(优思弗)口服。
2.6 皮肤科
患者皮肤表现包括两个方面:第一,眶周色素沉着斑伴眉周毳毛增多,该症状已出现近20年。患者既往无局部皮疹、外用糖皮质激素、特殊化妆品、护肤品及化学制剂等接触史。考虑以下疾病:(1)Becker’s痣:为一种表现为边界不规则色素沉着斑的错构瘤,常发生于躯干两侧上方,可伴毛发增多。该患者皮损位置不典型,暂不考虑该病。(2)局限性毛增多症:为多种遗传性皮肤病的主要或次要诊断特征。如阳光暴露部位的毛增多症可为卟啉病的症状之一,低前发际线和连眉可为Cornelia de Lange综合征的重要诊断依据。该患者起病晚,临床表现暂无法与某种遗传综合征相对应,暂不考虑遗传性疾病。迟发型卟啉病虽可成年起病,但典型皮损表现为暴光部位红斑水疱,可伴毛增多症。该患者无光暴露后出现典型红斑水疱史,卟啉病可能性小。(3)获得性局限性毛增多症:反复外伤、摩擦、刺激或炎症可致皮肤受累区域毛发变粗变长。该患者无既往皮疹史及特殊接触史,暂不考虑。(4)获得性系统疾病:部分青少年皮肌炎患者可出现髌下多毛症,机制尚不明。此外,毛增多症还可出现于胫前粘液性水肿、Rosai-Dorfman病等,但均较少见。(5)药物诱发的毛增多症:使用抗生素、糖皮质激素、米诺地尔、抗癫痫药、环孢素等药物可引起面部毛发增多,患者无上述用药史,暂不考虑。第二,腹背部皮肤暗紫红斑,腹部触诊质韧。结合病程中出现的呼吸无力、进食哽噎感及辅助检查,需考虑系统淀粉样变性、肌肉疾病以及结缔组织病。其腹壁皮肤活检低倍镜下表皮改变不明显。股四头肌活检刚果红染色真皮全层小血管周围未见红染物质沉积,真皮深部汗腺导管及腺腔部分红染,皮下脂肪组织小血管管壁极少量散在阳性,考虑为非特异性染色,而非淀粉样变典型病理表现。真皮及皮下组织小血管管壁增厚考虑为反应性,不具有诊断特异性。综合考虑患者的皮损特点和病理结果,目前淀粉样变证据不足。结合患者心脏、膈肌等多系统受累及ANA、抗AMA-M2、Ro52抗体阳性等结果,考虑眶周及腰背部皮肤改变为系统性疾病皮肤受累表现,建议进一步完善免疫性疾病、内分泌及代谢性疾病筛查。
2.7 康复医学科
该患者核心肌肉力量明显减弱,存在膈肌力量下降,呼吸模式欠佳,胸椎后凸,导致通气不足;同时存在心功能不全,二者是导致喘憋及活动耐力下降的最主要原因。患者的康复目标为减少并发症风险(主要为误吸和吸入性肺炎)、增加通气量、减少废用性肌肉力量下降,并进行姿势管理、核心肌肉力量与日常生活活动能力训练。减少并发症:建议给予均匀软糊状食物,同时进行口面动作训练指导。增加通气量:(1)呼吸模式指导:指导患者练习前后径扩张为主的腹式呼吸;(2)吸气肌力量训练:每天上下午各30次;(3)肺容量练习:对患者进行肺量计视觉指导和练习。减少废用性肌肉力量下降及姿势管理:(1)指导患者进行股四头肌姿势训练;(2)进行床旁腰背部肌肉姿势训练及背部伸肌姿势训练;(3)核心肌肉力量训练:通过起坐及伸髋练习,增加腹肌和臀肌等核心肌肉力量。日常生活活动能力训练:对于进食、穿衣、洗漱、翻身、起坐转移、站立等患者可以耐受的活动,鼓励其继续坚持。因患者在吸氧状态下才能维持稳定的氧合,暂不进行心肺耐力康复。
3 第一次多学科讨论后处理
予以托伐普坦(7.5 mg/d)、呋塞米(20 mg/d)、螺内酯(20 mg/d)以减轻心脏容量负荷,口服曲美他嗪改善心肌代谢(家属拒绝行心肌活检及右心漂浮导管检查)。低流量吸氧(2~3 L/min)并间断予以BiPAP辅助改善肺通气[4~6 h/d,模式S/T,IPAP为14 cm H2O(1 mm H2O=0.098 kPa),EPAP为4 cm H2O,FiO2为30%]。治疗20 d后喘憋症状未再发作,可平卧,下肢水肿基本消退。2020年11月17日起在康复医学科指导下每日进行床旁肺容量、核心肌肉力量及日常生活活动能力训练(15~20 min/次,1次/d),可耐受。口服优思弗(150 mg/次,3次/d),1周后复查GGT 337 U/L→240 U/L。2020年12月1日患者出院。出院前复查动脉血气分析,PaCO2下降至58 mm Hg。
2020年12月10日神经病理实验室肌肉病理结果回报:HE染色见肌纤维明显大小不等,部分肌纤维中重度萎缩,个别肌纤维肥大/变性/坏死、吞噬,未见再生肌纤维。肌内膜少数小血管周围可见单个核细胞浸润(图4A)。Gomori染色未见破碎红纤维等线粒体病典型表现。PAS染色未见糖原颗粒沉积,ORO染色未见明显脂滴沉积。COX染色示个别肌纤维膜下局部深染,肌纤维网状结构紊乱,肌纤维内斑片状淡染(图4B)。ACP及NSE染色见散在少数坏死肌纤维,其内粗大阳性颗粒增多。ATP染色示Ⅰ型和Ⅱ型肌纤维比例、分布大致正常,未见同型肌纤维群组化分布现象。进一步完善免疫组化染色,示肌内膜及坏变肌纤维内少量CD4+T细胞、CD68+T细胞浸润,未见CD8+T细胞及CD20+T细胞。MHC染色示肌纤维膜MHC-I表达增加(图4C)。抗C5b-9染色可见多数肌纤维周边阳性染色(图4D)。病理诊断:肌源性改变,结合免疫组化表现,考虑为免疫介导的炎性肌病。

图 4 患者股四头肌病理及免疫组化染色结果A.HE染色示肌纤维明显大小不等,部分肌纤维中重度萎缩,个别肌纤维肥大/变性(×100);B.COX染色示个别肌纤维膜下局部深染,个别肌纤维内斑片状淡染(×100);C.MHC染色示肌纤维膜有阳性着色(×100);D.抗C5b- 9染色示许多肌纤维周边阳性染色(×100)
4 第二次多学科讨论(2020- 12- 17)
4.1 风湿免疫科
结合患者肌肉病理及免疫组化结果,倾向于炎性肌病的诊断。在炎性肌病中,存在一种特殊类型的肌炎,即抗线粒体抗体相关炎性肌病(inflammatory myopathy associated with anti-mitochondrial antibodies)[5],临床表现为慢性骨骼肌受累和严重的心脏受累,包括心肌炎、心律失常和心肌病。该患者临床表现不典型,考虑为病程缓慢迁延所致。
4.2 心内科
神经病理结果结合患者临床表现符合炎性肌病合并心脏受累的诊断。回顾我院75例炎性肌病合并心脏受累病例的临床资料,40%合并心律失常(频发多源性室性早搏、短阵室性心动过速),且AMA阳性是合并室性心律失常的独立危险因素[6]。治疗方面,经前期针对心力衰竭的对症治疗,相关症状已明显改善。后续可考虑应用血管紧张素受体脑啡肽酶抑制剂治疗,随访观察其临床变化,并行超声心动图监测心功能变化。
4.3 皮肤科
该患者眶周色素沉着伴眉周毳毛增多,考虑为继发改变。自身免疫病或代谢性疾病均可继发色素沉着。自身免疫病累及皮肤多发生在真皮与表皮交界处,由于黑色素细胞分布于基底层,基底层受破坏后引起色素异常,表现为色素沉着或减退,亦称异色症。结合该患者病史及肌肉活检等结果,符合自身免疫病的诊断。其眶周色素沉着考虑为长期慢性自身免疫反应破坏皮肤基底层所致。
4.4 医学科学研究中心干细胞生物学平台
在某些慢性炎症中,细胞因子或炎症因子可通过激活黑素干细胞和黑素皮质素受体1(melanocortin 1 receptor, MC1R)/小眼畸形相关转录因子(microphthalmia-associated transcription factor, MITF)通路,刺激黑素细胞产生黑色素或褐色素[7],或可解释患者病程中出现的眶周色素沉着和眉周毳毛增多现象。患者肌肉、肺和皮肤病变在神经系统是否有共同病理特征,需进一步行遗传咨询。
5 第二次多科讨论后处理
继续予小剂量利尿剂以及曲美他嗪、β受体阻滞剂口服(剂量同前),家庭氧疗(每日低流量吸氧6~8 h),居家康复练习。2020年12月随访,患者喘憋未再发作,无下肢水肿,可室内活动并做简单家务。
6 最终诊断
抗线粒体抗体相关炎性肌病[心脏受累(射血分数保留的心力衰竭,Ⅰ度AVB,频发室性早搏,短阵室性心动过速);膈肌受累(Ⅱ型呼吸衰竭);皮肤系统受累(眶周色素沉着)];PBC;冠状动脉粥样硬化性心脏病(累及前降支、回旋支);高血压3级;2型糖尿病;高脂血症。
7 讨论
IIM是一组具有较高异质性的自身免疫性疾病,以进行性肌无力、肌电图异常、肌酶升高和肌肉炎症细胞浸润为特征,常伴有其他系统受累表现,如发热、皮疹、关节痛、间质性肺病、心肌病、吞咽困难等[8]。一般通过临床表现、相关抗体筛查、神经电生理、影像学及肌肉活检可明确诊断。早期的IIM特指PM和皮肌炎。1975年,Bohan等[2]提出经典IIM分型:包括原发性PM、原发性皮肌炎、合并血管炎的儿童PM或皮肌炎、合并肿瘤的PM或皮肌炎,以及合并其他结缔组织病的PM或皮肌炎。随着肌肉病理活检的开展及越来越多的抗体被检测发现,IIM分型更加细致。2018年,Selva-O’Callaghan等[9]提出新的成人IIM分型:其包括皮肌炎、免疫介导的坏死性肌病(immune-mediated necrotizing myopathy, IMNM)、散发性包涵体肌炎(sporadic inclusion body myosistis, sIBM)、重叠性肌炎(overlap myosistis, OM)和PM。有关肌炎的抗体包括肌炎相关性抗体(myosistis-related autoantibodies, MAAs)和肌炎特异性抗体(myosistis-specific autoantibodies, MSAs)[10]。
抗线粒体抗体相关炎性肌病是IIM中较罕见的由免疫介导的肌病类型。我国人群抗线粒体抗体相关炎性肌病患病率约为5.15%[11],与法国相近(7.80%)[12],但高于美国(0.6%)[5],低于日本(11.3%~19.5%)[13]。患者多于40~70岁发病,呈慢性病程,临床表现多样,可出现近端肌无力、轴向肌受累、翼状肩胛等[5,12- 14]。Albayda等[5]于2018年报道了7例抗线粒体抗体相关炎性肌病患者(1例皮肌炎、6例PM)的临床特征。3例合并其他自身免疫性疾病,包括PBC、自身免疫性肝炎、银屑病和桥本甲状腺炎。患者均有近端肌力下降,其中4例存在吞咽困难,5例合并心脏受累包括心律失常(传导阻滞、房性心动过速、室性期前收缩)及心肌炎(2例)。实验室检查均伴CK升高、AMA阳性,其中2例伴ANA阳性;肌电图示均存在肌源性损害;MRI示3例股四头肌水肿改变,2例存在脂肪浸润。我国学者[11]亦对该病的临床特征进行了总结。在收集的136例IIM患者中共检出抗线粒体抗体相关炎性肌病7例,包括皮肌炎3例、PM 2例、IMNM 2例。中位诊断年龄55.5(41,70)岁,发病至首次就诊时间为1~24个月。临床表现均有近端肢体无力,其中4例同时出现远端肌力下降及不对称肢体无力,4例伴肌痛,5例伴吞咽困难,3例伴构音障碍,1例伴呼吸困难。其他系统受累情况:2例伴心律失常(表现为传导阻滞和房性早搏),3例伴眶周紫色皮疹(均为皮肌炎患者),2例合并PBC,1例疑诊恶性肿瘤。辅助检查方面:6例CK升高;除AMA阳性外,2例抗NXP- 2抗体强阳性,1例抗T1F1γ抗体强阳性,3例存在大腿肌肉萎缩伴脂肪浸润。
本例患者慢性病程,多肌群受累,有明显的心脏受累,主要表现为心律失常及心力衰竭,AMA强阳性,合并PBC,虽CK无明显升高,但股四头肌活检补体免疫组化染色示抗C5b- 9染色及MHC染色均为阳性,符合炎性肌病的病理表现。此外,患者眶周色素沉着、眉周毳毛增多,可能由于慢性病程中,炎症因子刺激黑素细胞产生黑色素,以致眶周色素沉着。
治疗方面,一般予以激素治疗或激素联合其他免疫治疗,多数患者经治疗后肌力及CK水平明显改善。但AMA滴度无明显下降,故不建议使用AMA滴度评估疾病活动度[5,11- 13]。北京协和医院既往诊治的7例AMA相关炎性肌病患者中,均予以口服强的松1 mg/(kg·d),其中3例接受了其他免疫治疗。中位随访时间30.26个月,1例因恶性肿瘤去世,余6例临床症状均明显改善,CK均下降,但仍可检出AMA阳性[11]。本例老年患者,合并高血压、2型糖尿病等基础疾病,呈衰弱状态。经与患者及其家属沟通,综合风险与获益,暂未启动激素及免疫抑制治疗,给予纠正心力衰竭、改善二氧化碳潴留等对症治疗后,患者喘憋未再发作,下肢水肿消退。后续病情及转归需进一步观察随访。
患者病程中红细胞沉降率、超敏C反应蛋白等炎症指标正常,CK仅轻度升高,与IIM的诊断存在3个不匹配:(1)病程与症状不匹配:炎性肌病一般亚急性或急性发病,该患者病程8年,未进行诊治,不符合炎性肌病的病程进展;(2)CK酶指标与症状不匹配:已累及中轴肌(如呼吸肌)的炎性肌病患者CK往往显著升高,该患者CK仅间断轻度升高;(3)中轴肌受累与外周肌严重不匹配:炎性肌病一般表现为四肢近端肌无力起病,逐渐累及中轴肌,该患者主要表现为与外周肌不平行的核心肌肉力量明显减弱。因此,该患者临床表现很不典型,考虑为病程缓慢迁延所致,入院时已无急性活动性炎症。明确疾病诊断需从诸多症状及临床表现中梳理线索、厘清思路。该患者最终以膈肌运动减低引起的限制性通气功能障碍为线索,结合临床与影像学表现及股四头肌病理与免疫组化结果,得以确诊。该病例诊疗过程反映了多学科团队合力解决临床问题,并从临床问题中提出科学问题,进行转化医学探讨的重要价值。
8 专家点评
北京协和医院老年医学科 孙晓红教授
该患者老年女性,病情复杂,多系统受累,经两次多学科团队会诊,最终诊断为抗线粒抗体相关炎性肌病。经纠正心力衰竭、间断BiPAP治疗以及包括呼吸肌在内的康复训练,其临床症状明显改善。回顾整个诊疗过程,很多经验值得总结。
(1)以点及面,形成开阔的诊疗思路。患者以心功能不全、呼吸衰竭为主要表现入院,胸部CT提示膈肌上抬、食管扩张,以此为突破点,发现患者多肌群受累,完善肌活检及相关检查,从而明确诊断。
(2)多学科协作的重要性。该患者诊疗过程中,多学科团队共同参与和讨论,逐步厘清病因,解决诊疗决策中的难题,体现了多学科协作在疑难病诊疗中的重要作用。
(3)医患沟通及临床共同决策的意义。患者肌炎慢性化,目前无明显活动性炎症证据,后续是否应用激素和免疫抑制剂需审慎考虑。多学科讨论后与患者及家属进行充分沟通,鼓励患者继续居家进行康复练习,以增加通气量,减少废用性肌肉力量下降,并改善日常生活活动能力,医患沟通决策实现了患者受益最大化。
作者贡献:路菲负责病例整理、文章撰写及修订;张宁负责提供文章构思及修订;康琳、孙晓红、田然、郭潇潇、留永健、张文、彭琳一、苏童、曹剑、王怡宁、钱敏、曹宇泽、杨璐、刘跃华、张路、赵肖奕、冷泠参与多学科讨论并提供诊疗思路。
利益冲突:所有作者均声明不存在利益冲突
注:本文发表已获得患者本人知情同意。
猜你喜欢
中国比较医学杂志(2022年8期)2022-11-21
中西医结合心脑血管病杂志(2022年19期)2022-11-19
浙江农业学报(2022年1期)2022-11-07
汕头大学学报(自然科学版)(2022年3期)2022-08-31
中国现代医生(2022年21期)2022-08-22
西北民族大学学报(自然科学版)(2022年2期)2022-07-06
灌篮(2020年36期)2020-05-16
商情(2018年9期)2018-03-29
中国市场(2017年5期)2017-03-15
大众健康(2015年12期)2015-09-10
