
ATP7B基因复合杂合突变合并妊娠期急性脂肪肝1例报告
2022-02-11 09:39张翠薇邓明明
临床肝胆病杂志 2022年1期
杨 丹, 张翠薇, 邓明明
西南医科大学附属医院 a. 消化内科, b. 病理科, 四川 泸州 646000
1 病例资料
女性患者,28岁,因“停经39+6周,浮肿1个月余,血压升高2 d”于2020年9月20日入院。患者孕39+6周,正规产检。患者6年前及5年前分别顺产一女,新生儿体健,1年前人流一次,均无大出血史,体健。否认近亲婚配史及家族史。入院查体:血压142/91 mmHg,BMI 44.3 kg/m2,贫血貌,心肺未见异常,腹膨隆,腹壁浮肿,胎心率140 次/min,双下肢对称凹陷性水肿。入院后辅助检查:血糖3.2 mmol/L,AST 41.7 U/L,Alb 22.8 g/L,TBil 42.4 μmol/L,DBil 34.0 μmol/L,总胆汁酸24.2 μmol/L,GGT 482.8 U/L,ALP 590.1 U/L,凝血酶原时间14.1 s,D-二聚体5.17 μg/mL,纤维蛋白降解产物12.50 μg/mL,AFP 383.27 ng/mL。血常规、Coombs试验、红细胞形态检查、肾功能、自身免疫性肝病血清检测、肝炎病毒抗体、自身抗体谱均未见异常。腹部B超:亮肝,肝硬化,肝实质内查见多个强回声,最大约1.4 cm×1.4 cm。输入丁二磺酸腺苷蛋氨酸退黄,维生素K1、新鲜血浆纠正凝血异常,输入白蛋白,并急诊行剖宫产+子宫次全切除术终止妊娠。新生儿体健。术后患者出现高热,血浆引流管出现大量血液,2 h内超过3000 mL。复查:白细胞计数21.97×109/L,血红蛋白浓度54 g/L,血小板56×109/L,Alb 24.0 g/L,肌酐81.6 μmol/L,凝血酶原时间16.8 s,D-二聚体>20.00 μg/mL,纤维蛋白降解产物127.60 μg/mL。给予美罗培南抗感染,积极扩容,输注大量红细胞悬液、新鲜冰冻血浆及冷沉淀。患者于术后第2天出现意识丧失,血氧饱和度下降,查动脉血气分析示:pH 7.225,血二氧化碳分压65.9 mmHg,血氧分压52 mmHg。立刻气管内插管,利尿,营养支持,继续输血。治疗期间患者右上肢肿胀明显,血管超声提示:右侧头静脉前臂段血栓形成,予以依诺肝素钠皮下注射抗凝。经治疗后患者情况好转。出院诊断:(1)妊娠期急性脂肪肝(acute fatty liver of pregnancy,AFLP);(2)肝硬化;(3)肝内再生结节;(4)失血性休克;(5)弥散性血管内凝血;(6)妊娠期高血压疾病;(7)肥胖症;(8)G4P3 40+1周剖宫产+子宫次全切除术后;(9)右侧头静脉前臂段血栓形成。嘱患者继续口服熊去氧胆酸和利伐沙班。
2个月后患者因“关节痛6个月余,腹胀8 d”再次入本院消化内科。复查:AST 45.6 U/L,Alb 35.0 g/L,TBil 38.4 μmol/L,DBil 21.9 μmol/L,TBA 20.1 μmol/L,GGT 115.8 U/L,ALP179.9 U/L,AFP 12.00 ng/mL,凝血检查正常。入院时查体:血压100/74 mmHg,BMI 21.8 kg/m2,眼底未见Kayser-fleischer环,移动性浊音阳性,右膝关节肿胀。进一步完善检查:Coombs试验阳性, 铜蓝蛋白 0.038 g/L,24 h尿铜525 μg。肌骨超声检查:右膝关节积液,滑膜增生。腹部CT血管造影未见异常征象。肝静脉压力梯度13.5 mmHg。肝穿刺活检(图1):考虑肝豆状核变性(hepatolenticular degeneration,HLD)。铜转运P型ATP酶(copper-transporting P-type ATPase, ATP7B)基因测序:ATP7B p.A778 L,p.L1188P复合杂合突变(图2),最终诊断为HLD。嘱患者口服曲恩汀,3个月后复查肝功能,恢复正常。
2 讨论
HLD是一种由ATP7B基因突变所致铜代谢障碍性常染色体隐性遗传病,我国较欧美多见,人群发病率为3/10万,携带者的比例为1∶90,女性多于男性[1-2]。ATP7B是具有多个结构域的跨膜蛋白,在肝细胞的高尔基体上集中表达。游离铜在ATP7B的作用下形成铜蓝蛋白由肾脏代谢;或者由ATP7B转运至溶酶体,以囊泡的形式依赖胆汁排泄[3]。当ATP7B基因发生突变后,血铜排泄受阻,异常沉积在肝脏、角膜、肾脏和基底节,导致相应脏器损害。目前已经发现的ATP7B基因突变位点接近800个,各突变位点与临床表现并无确切的对应关系,且患者可能存在多个复合突变,这就导致HLD的临床表现复杂,起病隐匿。如果在肝铜蓄积期就及时确诊,患者大多预后良好,如果以急性暴发性肝衰竭起病,则预后不佳[4]。东亚人群最常见的突变类型是ATP7B p.R778L,临床表现以肝损伤为主[5]。本例患者为ATP7B p.A778L,p.L1188P复合杂合突变,其主要的致病突变为p.A778L,功能研究显示该突变可降低细胞内铜离子的外排作用且影响ATP7B蛋白在细胞内的定位和运输[6]。多数研究认为HLD携带者症状轻微,生化检查轻度异常,但像本例患者一样急性暴发性肝衰竭起病,并在短期内发展为肝硬化较为罕见。是什么原因导致患者在孕期短时间出现暴发性肝损伤呢?

注:a,可见大泡性脂肪变性,部分可见空泡核、糖原核(HE染色,×200);b,汇管区纤维增生和淋巴细胞浸润,假小叶形成(HE染色,×200);c,铜染色阳性(×200);d:铁染色阴性(×200)。图1 患者肝脏穿刺活检病理
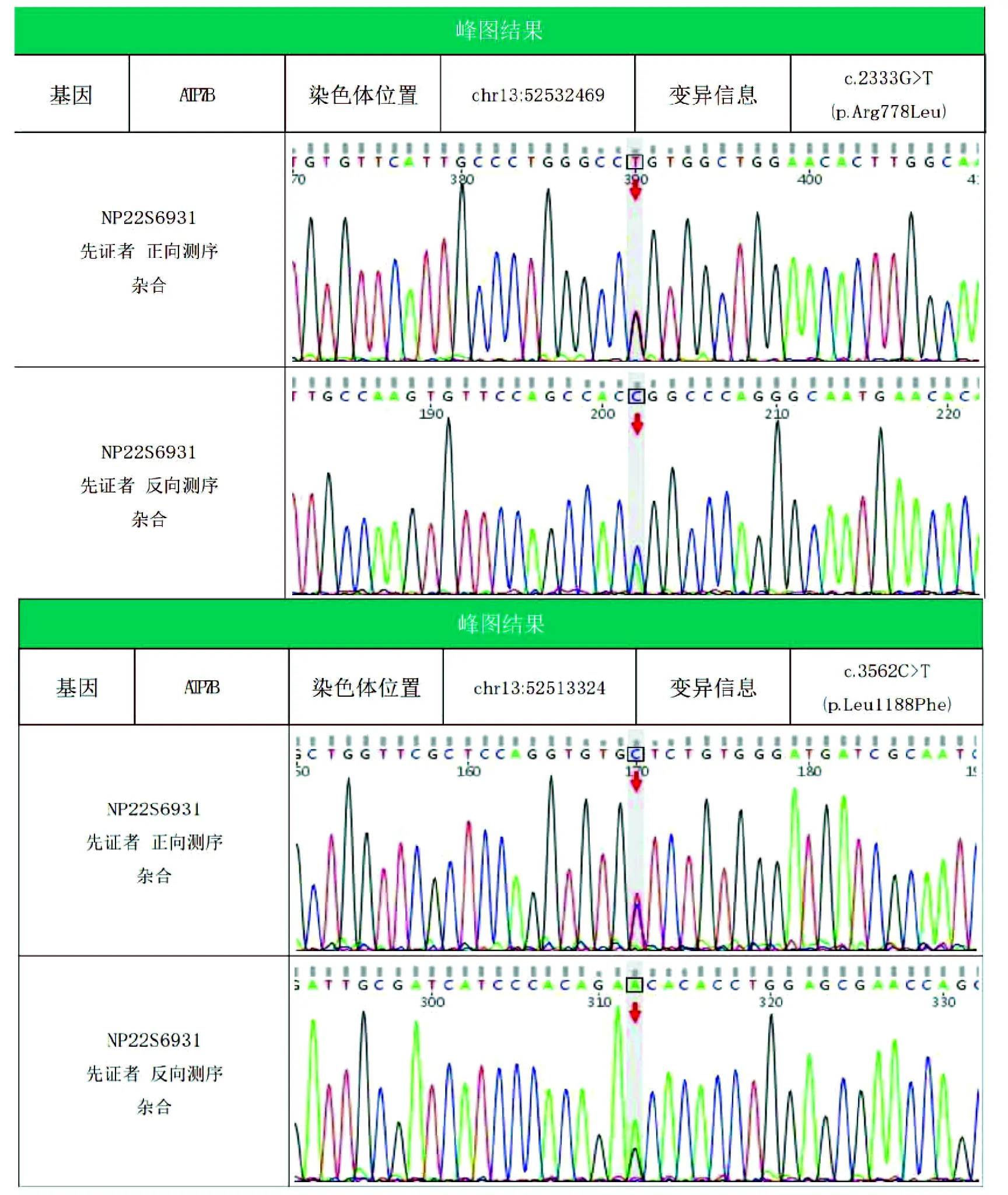
图2 患者ATP7B基因检测结果
ATP7B突变基因携带者的临床表型受多种因素的影响,除了突变类型、种族、年龄之外,饮食和脂质代谢也是较为重要的影响因素。HLD患者的肝脏病理中胆汁淤积和脂肪变性均十分常见,表明体内铜稳态受损与肝脂肪变性密切相关。研究[7]发现,HLD模型中固醇调节因子结合蛋白-1表达下调,抑制胆固醇合成,促使甘油三酯在肝细胞内沉积。这也进一步解释了为什么在本例患者的肝脏病理组织中可以看见大泡性脂肪变性。一般来讲,HLD患者都被建议避免食用富含铜的食物,而对饮食中糖、脂肪含量较少限制。研究[8]发现,喂养高热量饮食的HLD大鼠模型线粒体内铜的积累增加,结构严重破坏,导致过氧化氢生成增多和ATP功能异常,这都导致了HLD的较早发作并加速了疾病进展。本例患者在孕期进食大量高热量、高脂饮食,缺乏运动,孕晚期BMI高达44.3 kg/m2,同时合并了AFLP,而AFLP发病机理与怀孕期间脂肪酸代谢缺陷有关,患者和胎儿均可能存在遗传缺陷[9],这些都可能改变患者的表观遗传,导致患者孕晚期出现爆发性的肝衰竭。一例单卵HLD双胞胎患者的病例报告[10]指出,患有神经性贪食症的妹妹出现了明显的肝衰竭,被迫接受了肝移植,而长期处于营养不足的姐姐则仅表现为无症状性肝功能异常。在本病例中,患者术后通过减轻体质量,改变饮食结构,2个月后复查肝功能仅轻度异常,凝血恢复正常,提示减轻体质量、改变饮食可能改善HLD患者病情,但机制尚待研究。孕晚期由于水钠潴留、蛋白质需求增多、饮食结构严重失衡(蛋白质摄入不足)、肝功能异常(蛋白质合成障碍)等多重因素作用导致患者在产前出现与肝功能损害不平行的严重低白蛋白血症。血浆纤维蛋白原合成减少、病理产科、剖宫产等都可能导致患者术后出现弥散性血管内凝血,大出血。因此,对患有妊娠期肝病的患者可能需要更为积极地处理低蛋白血症和预防弥散性血管内凝血发生、发展。
目前对HLD治疗的专家共识是:诊断后应立即开始治疗,即使在处于前驱期,也应终生治疗;应定期进行肝脏和血液学检查,神经系统检查和铜代谢情况,相应地调整治疗方案[11]。因为涉及终生治疗,所以依从性和安全性是选择药物必须要考虑的关键问题。目前首选的一线用药是曲恩汀[12],推荐剂量最初为20 mg/kg,以后为15 mg/kg,可每日一次给药[13]。锌盐是不能耐受螯合剂患者的替代疗法,也是无症状或稳定患者的初始和维持疗法,对幼儿及孕妇目前也认为是安全的[14]。但长期使用锌盐可能导致缺锌和低铜相关性疾病,需密切检测血锌和血铜的浓度[15]。
综上所述,本例HLD患者为ATP7B p.A778L,p.L1188P复合杂合突变,以AFLP、肝硬化、关节炎为主要临床表现,终止妊娠及控制体质量后病情好转,通过肝组织病理检查和基因检测确诊,曲恩汀治疗效果良好。对于HLD患者,尤其在妊娠期,要加强饮食指导,除了限制高铜饮食以外,也应限制脂肪和热量的摄入。再次强调对于不明原因肝损伤、关节炎要及时想到HLD的可能,认真询问家族史和遗传史,即使均无异常也不能大意,可完善血清铜、铜蓝蛋白和24 h尿铜排除。曲恩汀是HLD的首选药物,依从性和安全性均高于青霉胺。临床医生应通过本病例加深对HLD认识,早期确诊并选择适宜的治疗方案,避免肝铜蓄积,预防急性暴发性肝炎,减少死亡和降低医疗成本。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:杨丹负责拟定研究思路,撰写论文;张翠薇负责收集分析数据;邓明明负责修改论文和最终定稿。
猜你喜欢
中老年保健(2022年6期)2022-11-25
军事文摘(2022年12期)2022-07-13
中国新时代 (2022年4期)2022-04-14
医学前沿(2021年18期)2021-04-14
初中生学习指导·提升版(2020年5期)2020-09-10
祝您健康(2019年8期)2019-08-09
新教育时代·教师版(2017年30期)2017-09-12
家用汽车(2016年9期)2016-11-04
数理化学习·高三版(2015年3期)2015-10-21
职业·中旬(2009年12期)2009-06-01
