
胆汁淤积性肝病管理指南(2021)
2022-02-11 09:39中华医学会肝病学分会
临床肝胆病杂志 2022年1期
中华医学会肝病学分会
胆汁淤积是指肝内外各种原因造成胆汁形成、分泌和排泄障碍,胆汁流不能正常流入十二指肠而进入血液的病理状态,临床可表现为瘙痒、乏力、尿色加深和黄疸等,早期常无症状,仅表现为血清ALP和GGT水平升高,病情进展后可出现高胆红素血症,严重者可导致肝硬化肝衰竭甚至死亡[1-2]。各种原因使肝脏病变导致胆汁淤积为主要表现的肝胆疾病统称胆汁淤积性肝病,胆汁淤积本身也会进一步加重肝脏的损害。
为帮助临床医生规范诊治胆汁淤积性肝病,中华医学会肝病学分会、中华医学会消化病学分会和中华医学会感染病学分会2015年组织国内有关专家制定了《胆汁淤积性肝病诊断和治疗共识》[3]。近年来随着我国胆汁淤积性肝病临床资料的不断积累和完善,在原先共识的基础上进行了更新形成本指南。本指南主要介绍胆汁淤积性肝病的病因、分类、临床表现、诊断标准、治疗原则、遗传和妊娠胆汁淤积性肝病、胆汁淤积肝外表现诊断和处理。药物性、酒精性、乙型肝炎、丙型肝炎、原发性胆汁性胆管炎(primary biliary cholangitis,PBC)、原发性硬化性胆管炎(primary sclerosing cholangitis,PSC)、自身免疫性肝炎和代谢相关脂肪性肝病等所致的胆汁淤积诊断和治疗可参照相应的指南。指南中提及的证据和推荐意见基本按照GRADE系统进行分级(表1)。
1 病因和分类
胆汁由肝细胞和胆管细胞产生,人每天总胆汁流约600 mL,肝细胞提供胆盐依赖性胆汁(约225 mL/d)和非胆盐依赖性胆汁(约225 mL/d),胆管细胞提供另外150 mL/d的胆汁。胆汁淤积是胆汁流量或形成障碍[4]。胆汁淤积性肝病按发生部位可分为肝内胆汁淤积和肝外胆汁淤积。肝细胞功能障碍或毛细胆管、细胆管(<15 μm,亦称闰管或Hering管)及小叶间胆管(15~100 μm)病变或阻塞所致胆汁淤积称肝内胆汁淤积[5-7];在胆管影像学上没有主要胆管堵塞的表现,主要原因有药物、酒精、病毒、细菌和免疫等。间隔胆管(>100 μm)、区域胆管(300~400 μm)、 节段胆管(400~800 μm)、 左右肝管、胆总管至壶腹部的病变或阻塞所致胆汁淤积称肝外胆汁淤积[2,7]。肝外胆汁淤积的条件是胆管堵塞,通常在肝外,但生长在肝内胆管肝门胆管癌也包括在内。常见原因是胆管结石,胰腺、胆道和壶腹部癌,良性胆道狭窄。通常这些原因引起的是急性胆汁淤积[8]。如胆汁淤积持续超过6个月,则称为慢性胆汁淤积[5]。区分肝外和肝内胆汁淤积很重要,单凭症状和体征及生化改变可能不能区分,需要系统诊断来鉴别。

表1 推荐意见的证据等级和推荐强度等级
原发性硬化性胆管炎(PSC)可累及小和大肝内胆管和/或肝外胆管,因此部分患者可同时有肝内和肝外部分病变(图1)。
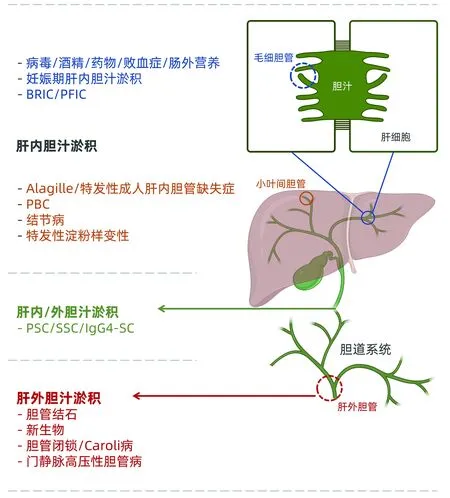
图1 胆汁淤积性肝病与胆道系统发生部位
根据细胞学损害部位又分为肝细胞性和胆管细胞性[5],肝细胞和胆管细胞均有损害的称混合性胆汁淤积。
2 流行病学
胆汁淤积性肝病的发生率目前尚无确切数据。Bortolini等[9]即对初次诊断慢性肝病患者研究显示2520例初次诊断慢性肝病患者中882例(35%)出现胆汁淤积,胆汁淤积更易出现在PBC和PSC。1000例慢性病毒性肝炎患者研究[10]显示,56%患者出院时ALP或GGT仍然高于正常值上限(ULN),且这些指标异常的患者中肝纤维化和肝硬化的发生风险和病情严重程度显著增加。曹旬旬等[11]基于上海市4660例住院慢性肝病患者调查结果显示胆汁淤积总发生率为10.26%,慢性肝病患者胆汁淤积发生率随年龄增加有上升趋势。
3 临床表现
除引起胆汁淤积原发疾病相关临床症状外,肝脏胆汁淤积本身可引起相关临床症状,以及因胆汁淤积而致的继发性改变。患者早期可无不适症状,可有乏力、纳差、恶心、上腹不适等非特异症状,胆汁淤积相关的临床表现主要有黄疸、皮肤瘙痒、疲劳、脂肪泻、黄色瘤和肝性骨营养不良等。
4 生物标志物
胆汁淤积诊断最常用的生物标志物包括ALP、GGT、胆汁酸和胆红素等。
4.1 ALP和GGT ALP和GGT升高是胆汁淤积最具有特征性早期表现。当胆汁排泄不畅,毛细胆管内压升高,可诱发ALP产生增多,加之胆汁酸凭借其表面活性作用,将ALP从脂质膜上溶析下来,使血清ALP明显升高[12]。胆汁淤积时ALP如何进入血液和升高的机制目前还不清楚。ALP升高除见于胆汁淤积外,也可见于妊娠、儿童生长期、骨骼疾病及部分肿瘤。GGT升高比其他血清酶出现得更早,持续时间更长,在肝脏酶中敏感性最高,但其特异性却比较低。GGT对胆汁淤积诊断灵敏性和特异性不低于甚至优于ALP。在排除酗酒等其他肝损伤情况下,若ALP和GGT同时升高,可确认存在肝细胞和胆管细胞损伤。若GGT升高而ALP不升高,几乎也可判定存在肝毛细胆管和胆管上皮细胞损伤。ALP升高病例,如果不合并有GGT升高,常可排除肝源性疾病。需要注意的是在一些特殊胆汁淤积性肝病如家族性肝内胆汁淤积(familial intrahepatic cholestasis, FIC)1、2、4、5、6型和USP53缺陷病等,表现为结合胆红素或胆汁酸升高,GGT可不升高[13-14]。
4.2 胆汁酸 胆汁酸对于诊断胆汁分泌受损较胆红素敏感,但是对于大多数的胆汁淤积不如ALP敏感,而且许多肝病如肝硬化、急慢性肝炎均可有胆汁酸升高。正常胆汁酸值在空腹时为1.0~6.0 μmol/L,餐后2 h为6.0~9.0 μmol/L。胆汁淤积时胆汁酸值超过10 μmol/L。胆汁酸值在10~20 μmol/L为轻度升高,20~40 μmol/L为中度升高,40 μmol/L以上为重度升高[1,4]。胆汁酸及甘胆酸虽然是反映胆汁淤积敏感指标,但检测方法学缺乏标准化,加上干扰因素多、特异性欠佳等因素,是目前国内外相关指南中未将其列入判断标准的重要原因。胆酸水平升高对肝胆疾病有特异性,但在确定各种原因所致的肝细胞损伤时,胆酸的敏感性比最初预期小。在空腹胆酸基础上测定餐后2 h胆酸也不能增加敏感性。此外,测定每种胆酸没有诊断价值[4]。
4.3 胆红素 胆汁淤积和肝细胞病变可引起胆红素升高,以直接胆红素升高为主,肝细胞损害引起的黄疸因为同时有摄取、结合、排泄的障碍,因此直接和间接胆红素均可升高,但一般直接胆红素升高比间接胆红素升高的幅度大。单纯胆红素升高(无肝酶升高)需要考虑是遗传性因素或血液系统疾病所致。
4.4 分子标志物 遗传性胆汁淤积性肝病与基因变异相关。传统的测序通过基于患者表型的特定基因的直接测定来确定。目前二代测序技术在临床得到应用,遗传相关的胆汁淤积性肝病如FIC更容易被诊断。表2总结了遗传性胆汁淤积性肝病的相关突变基因[15]。单基因肝病仅占肝脏疾病的一小部分,更多的是多基因或基因和环境因素相关作用导致。
5 病理学
胆汁淤积时大体标本呈黄绿色,穿刺标本呈散在绿色斑点或通体深绿色。肝内胆汁淤积的基本病理变化是胆汁从肝小叶第3区肝细胞开始,表现为肝细胞内胆汁淤积,肝细胞呈羽毛状变性,伴毛细胆管扩张胆栓形成[6-7,16]。严重时以扩张含胆栓的毛细胆管为中心,肝细胞呈腺泡样排列,形成胆汁花环,这是肝内胆汁淤积的特征性病理变化。可见肝窦内增生肥大的Kupffer细胞吞噬胆汁,门管区小叶间胆管胆汁淤积伴胆栓形成。电镜观察显示毛细胆管微绒毛水肿、变短,直至消失。肝外阻塞性胆汁淤积组织病理学特征为门管区周边肝内胆汁湖伴胆汁肉芽肿形成,长期肝外阻塞可引起肝内继发性胆汁淤积。胆汁淤积的后期可引起门管区纤维化,甚至胆汁性肝硬化。
6 诊断
6.1 诊断标准 目前有关胆汁淤积性肝病的诊断标准和具体的指标尚未统一,以ALP和GGT作为诊断指标尚有一些争议。2009年欧洲肝病学会胆汁淤积性肝病处理临床实践指南专家诊断工作组[5]建议“ALP>1.5×ULN,且GGT>3×ULN”诊断胆汁淤积性肝病。2015年中华医学会肝病学分会《胆汁淤积性肝病诊断和治疗共识》[3]建议“ALP>1.5×ULN,且GGT>3×ULN”诊断胆汁淤积性肝病,但需注意在一些特殊胆汁淤积性肝病如“PFIC 1和2型及BRIC,GGT可不高。”鉴于现状、近年进展和认识,本指南推荐的胆汁淤积诊断标准为:ALP>1.5×ULN,且GGT>3×ULN。需注意在一些特殊胆汁淤积性肝病如FIC 1、2、4、5、6型和USP53缺陷病等,表现为结合胆红素或胆汁酸升高,GGT可不高。IFC 3型、Alagille综合征、Citrin缺陷病、胆管板发育畸形(Caroli病和先天性及囊性肝纤维化)和Niemann-Pick病(C1/C2型)等GGT很高。

表2 胆汁淤积性疾病累及基因
6.2 诊断步骤 首先通过血清学检测确定胆汁淤积是否存在;接着影像学和内镜等方法确定肝内和肝外胆汁淤积;最后综合分析得出诊断(包括病史、症状和体征、血液生化、影像学、内镜、肝活组织学和相关基因检测等)(图2)。
6.3 与黄疸区别和联系 胆汁淤积和黄疸不完全等同,胆汁淤积包括胆红素在内的全部胆汁成分淤积[1]。黄疸是血液胆红素浓度升高,皮肤和巩膜等处发生黄染的现象。胆汁淤积早期,仅有ALP和GGT升高,可不一定出现黄疸,只有当胆红素超过34.2 μmol/L临床上才显现黄疸。有些疾病仅有胆红素代谢障碍,而胆汁其他成分正常,如遗传性高胆红素血症(Gilbert综合征、Crigler-Najjar综合征、Dubin-Johnson综合征和Rotor综合征等),这些患者仅有胆红素升高,而ALP和GGT及胆汁酸并不升高。在一些溶血性疾病如遗传性球形细胞增多症、地中海贫血、阵发性睡眠性血红蛋白尿、获得性溶血性贫血和新生儿溶血病等溶血发生时,也会出现黄疸,但肝酶不升高。因此黄疸者需排除遗传性因素和溶血性疾病。
推荐意见1:肝生化检查发现ALP>1.5×ULN,且GGT>3×ULN可诊断胆汁淤积性肝病。(B1)部分FIC表现为结合胆红素和/或胆汁酸升高,GGT可不高。(B2)
推荐意见2:影像学检查是区分肝内和肝外胆汁淤积的主要临床手段,可根据情况选择腹部超声、CT和MRCP。(C1)
推荐意见3:常规影像学检查不能明确诊断,且临床高度怀疑肝外胆道梗阻或胆管炎时建议ERCP或超声内镜检查。(B1)
推荐意见4:对于无法解释的肝内胆汁淤积且AMA/AMA2和/或SP100和/或GP210阴性者应行其他抗体检测,以排除系统性或其他器官自身免疫性疾病,仍不能确定时可行肝穿组织学检查。(C1) 怀疑遗传性胆汁淤积症不能确诊者可检测相关基因。(B1)
推荐意见5:胆汁淤积性肝病早期可不出现黄疸,黄疸者需鉴别遗传性高胆红素血症和溶血性疾病。(B1)
7 治疗
7.1 治疗原则 治疗原则是去除病因和对症治疗。最有效治疗是病因治疗,如手术或经内镜取结石或手术切除肿瘤解除梗阻,PBC和PSC可用熊去氧胆酸(UDCA),药物性和酒精性肝病及时停用有关药和戒酒最为重要,乙型和丙型肝炎进行相应的抗病毒治疗,自身免疫性肝炎可用皮质激素和/或免疫抑制剂取得缓解,代谢相关脂肪性肝病通过饮食、运动和生活方式干预等。
7.2 药物治疗 药物治疗目的是改善由于胆汁淤积所致的临床症状和肝脏损伤。主要的药物有UDCA、S-腺苷蛋氨酸(S-adenosyl-L-methionine,SAMe)、考来烯胺、奥贝胆酸和贝特类药等[17-19]。
7.2.1 UDCA 治疗胆汁淤积疾病基于通过亲水性的、有细胞保护作用和无细胞毒性的UDCA来相对地替代亲脂性、去污剂样的毒性胆汁酸,以及促进肝细胞的分泌作用和免疫调节来完成。可用于治疗PBC、PSC、ICP、囊性纤维化、肝移植后淤胆、药物性胆汁淤积、FIC和Alagille综合征等。UDCA一般剂量为10~15 mg·kg-1·d-1,Byler病和Alagille综合征剂量可增至45 mg·kg-1·d-1,囊性肝纤维化剂量为20~25 mg·kg-1·d-1。
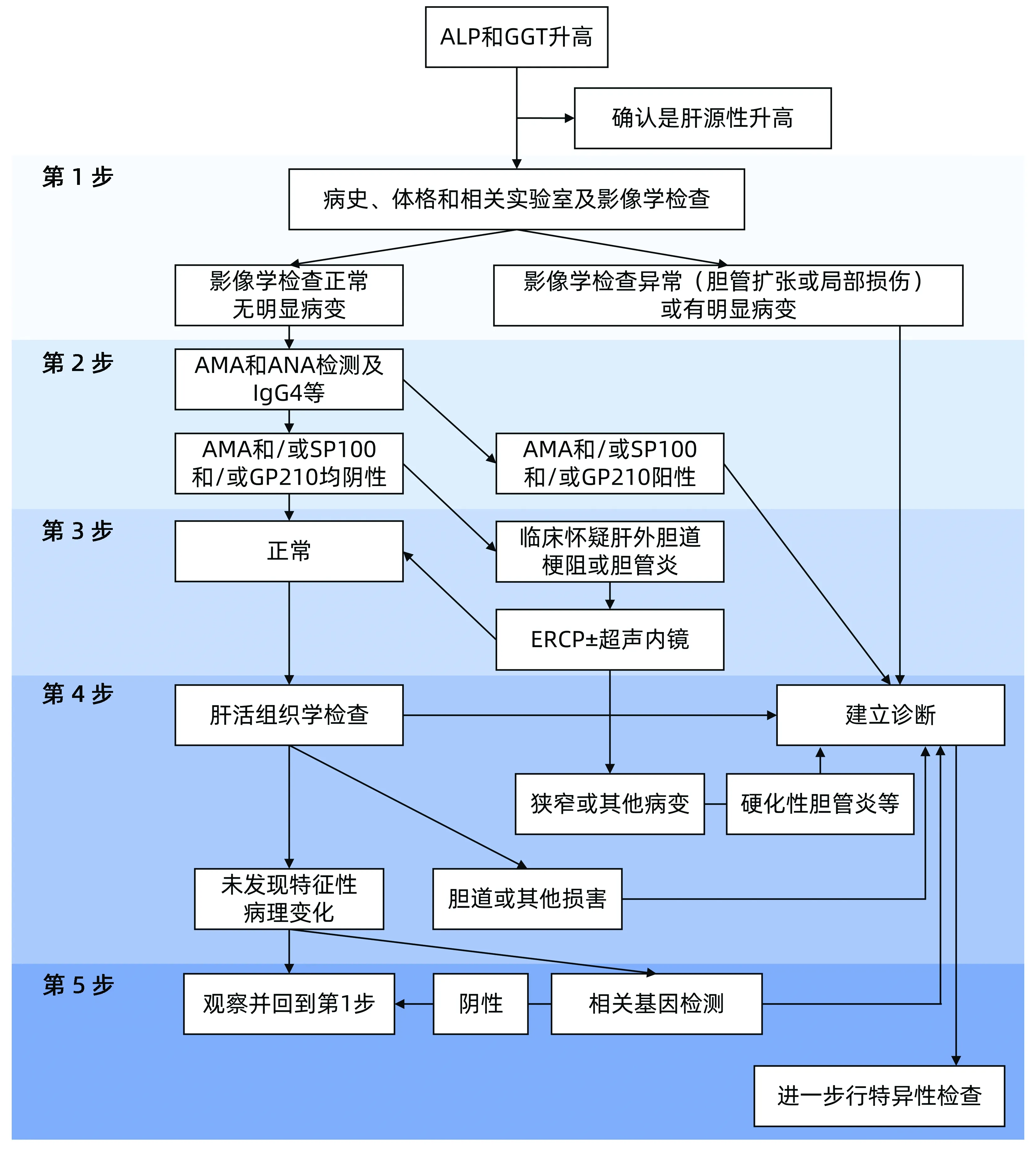
图2 胆汁淤积性肝病诊断流程图
7.2.2 SAMe 作为甲基供体(转甲基作用)和生理性巯基化合物(如半胱氨酸、牛磺酸、谷胱甘肽和辅酶A等)的前体(转硫基作用)参与体内重要生化反应。SAMe可用于肝细胞性胆汁淤积、ICP和药物性胆汁淤积。初始治疗,SAMe,0.5~1.0 g/d,静脉滴注;维持治疗,口服SAMe片,1.0~2.0 g/d。
7.2.3 考来烯胺 阴离子交换树脂,口服后在肠道与胆酸结合后随粪便排出,可使胆酸排出量比正常高3~4倍。口服12~16 g/d,分3次于饭前或睡前用水或饮料拌匀服用。与UDCA和其他药物服用的间隔至少在4 h以上。
7.2.4 奥贝胆酸 属FXR激动剂,通过FXR间接抑制细胞色素7A1(CYP7A1)基因表达,抑制胆酸合成,主要用于治疗对UDCA无应答的PBC。口服5~10 mg/d。
7.2.5 贝特类药物 过氧化物酶体增殖体活化受体(PPARs)激动剂,PPARs激活后可通过抑制胆汁酸合成酶CYP7A1的表达抑制胆汁酸合成,也可通过上调胆汁酸转运体多耐药蛋白3(MDR3)表达增加胆汁的排泌。口服非诺贝特 160~200 mg/d或苯扎贝特 (400 mg/d)。
7.2.6 其他治疗 有免疫机制介导的胆汁淤积者充分权衡治疗获益后酌情考虑应用糖皮质激素或免疫抑制剂,也可考虑应用紫外照射、体外白蛋白透析及鼻胆管引流等方法。胆汁淤积性肝病患者经积极内科治疗无效且6~12个月内可能死亡或MELD≥15应行肝移植评估。中医中药如茵栀黄、苦黄等对胆汁淤积性肝病有一定治疗作用,但尚需要临床积累更多的疗效和安全性数据。尚有许多新型药物正处于研发中包括FGF19类似物、norUDCA、Simtuzumab、Infliximab和粪菌移植等,有望在将来为胆汁淤积性肝病提供新的治疗方法。
推荐意见6:胆汁淤积性肝病治疗原则是去除病因和治疗胆汁淤积。治疗药物主要有UDCA(A1)、S-腺苷蛋氨酸(B1)、考来烯胺(B1)、贝特类(B1)和奥贝胆酸(B1)等,可单用或联用。
推荐意见7:经上述药物治疗无效者可酌情选用激素和/或免疫抑制剂、紫外线照射、体外白蛋白透析及鼻胆管引流等方法。(C2)
推荐意见8:胆汁淤积性肝病患者经积极内科治疗无效且6~12个月内可能死亡或MELD≥15应行肝移植评估。(B1)
8 遗传性胆汁淤积性肝病
8.1 囊性纤维化相关的肝病(cystic fibrosis-associated liver disease,CFLD) CFLD是一种常染色体隐性遗传性疾病,第7染色体长臂上的跨膜转导调节蛋白基因突变引起。表现为肝肿大、肝生化和超声检查异常,可伴有先天性胆汁淤积、肝脂肪变、局灶或多叶性肝硬化[20-21]。(1)诊断:CFLD尚未有明确诊断标准。1/3患者有肝肿大,其可由CFLD或肺心病肝充血所致。ALP、ALT、AST、胆红素和GGT持续异常超过1.5×ULN时应当进一步检查以严格评估肝损伤(凝血酶原时间、白蛋白)并排除其他原因的肝病。超声检查发现CFLD征象如肝肿大或胆道异常,肝脏内见数量及大小不一的无回声占位病变。腹部CT可显示囊肿的大小及残存多少正常肝组织。由于许多患者有局灶性纤维化、肝硬化的存在,肝穿组织学检查意义不大。(2)尚无对CFLD有益的治疗药物。予以20~30 mg·kg-1·d-1UDCA显示可持续改善肝生化指标、刺激受损胆管胆汁的分泌、改善组织学指标(2年以上)和营养状态。该病患者一般寿命长,预后常取决于同时存在的肾囊肿病的严重程度,癌变极少。本病很少需要手术治疗,有急性症状时可在超声指导下穿刺抽液,但囊液可重新产生。当患者日常生活严重受限或者终末期患者可考虑肝移植[20-22]。
推荐意见9:CFLD诊断基于肝囊性纤维化、肿大、生化指标异常及影像学检出肝脏内数量及大小不一的囊性病变。(C2)UDCA 20~30 mg·kg-1·d-1可改善CFLD肝生化和组织学指标。(C1)日常生活严重受限或者终末期患者行肝移植评估。(B1)
8.2 FIC FIC是一组常染色体隐性遗传疾病,分别由ATP8B1、ABCB11、ABCB4、TJP2、NR1H4、MYO5B及USP53基因突变,直接或间接导致肝细胞毛细胆管转运体异常所致,男女发病率无差异[8,13-14]。该病以肝内胆汁淤积性黄疸伴严重瘙痒为最常见表现,瘙痒甚至可严重影响生活质量,查体可见明显的皮肤抓痕,胆管造影术显示肝内或肝外胆管通畅。该病多表现为连续的疾病谱,从“良性”BRIC到严重型PFIC,肝组织学表现肝细胞或毛细胆管内胆汁淤积,多不伴胆管增生,可显示肝纤维化,最终发展为肝硬化和肝衰竭;部分病例可呈反复发作的自限性严重瘙痒和胆汁淤积,每次发作可持续数周至数月,然后有数月或数年的无症状,因此称为BRIC。BRIC在发作时肝组织学表现肝细胞或毛细胆管内胆汁淤积,但无明显纤维化;发作期间肝组织学和肝功能正常。最初认为尽管每次BRIC发作很严重,但不会发生进行性肝损伤和肝硬化,然而后来观察到一些所谓的BRIC病例反复发作,同样会进展为终末期肝病。因此目前有提出将“良性”这一词语删除[4]。也有作者观察到一些FIC病例仅在婴儿期出现胆汁淤积症,无反复发作,叫做暂时性婴儿胆汁淤积症[23]。
由ATP8B1、ABCB11、TJP2、NR1H4、MYO5B及USP53基因突变引起的FIC临床上以低GGT为特征,常从新生儿期起病,血中胆红素和胆汁酸明显升高,而GGT升高不明显(常<100 U/L)[13-14]。ATP8B1缺陷病,曾称为FIC 1型,包括PFIC 1型和BRIC 1型,致病机制尚不完全清楚。PFIC 1型又称作Byler病,除胆汁淤积症表现外,还可有腹泻、胰腺炎、发育障碍、听力缺失、甲状腺功能低下等肝外表现。电镜检查显示毛细胆管内有粗颗粒胆汁(Byler胆汁)。ATP8B1缺陷病的基因型和表型关系不密切[24]。ABCB11缺陷病,曾称为FIC 2型,包括PFIC 2型和BRIC 2型。PFIC 2型既往称作“Byler综合征”。ABCB11基因编码胆盐输出泵(BSEP),是目前已知人类肝脏毛细胆管膜上唯一负责将胆盐转运进入毛细胆管的转运泵,其缺陷造成胆汁酸蓄积在肝细胞内,引起肝脏炎症和肝细胞巨细胞变。PFIC 2型发生肝癌的风险较大,也可发生胆结石。基因型和表型关系密切[25-26]。TJP2基因编码紧密连接蛋白2,参与上皮细胞间和内皮细胞间连接的结构。TJP2缺陷病,又称FIC 4型,严重病例肝组织免疫组化可见TJP2蛋白表达缺失,常导致死亡或需要肝移植才能长期存活。新近发现该病也有明显的基因型表型关系,临床上也可表现为轻重不等的连续表型[27]。胆汁酸合成、分泌与代谢受到核受体家族蛋白的精细调节,其中FXR接收胆汁酸信号激活并进行反馈调节,是维持体内胆汁酸稳态最重要的蛋白。FXR由NR1H4基因编码,NR1H4缺陷病又称FIC 5型。可表现为严重的新生儿胆汁淤积及早发型非维生素K依赖型凝血障碍,迅速发展至终末期肝病,需要早期肝移植才能存活。MYO5B参与细胞内物质运输,在小肠上皮细胞微绒毛面形成、肝细胞毛细胆管面形成过程中至关重要。该基因缺陷有密切的基因型表型相关性,功能完全缺失引起微绒毛包涵体病,该基因功能部分缺失通过一种特殊毒性负性作用导致BSEP不能正确定位而致病,表现为轻重不等的胆汁淤积症谱系,也称FIC 6型[28]。USP53编码泛素特异性肽酶53,和TJP2有相互作用。USP53缺陷病除有胆汁淤积外,可伴有听力障碍,严重者会双耳失聪,电镜下观察到肝细胞间的紧密连接结构变长[29]。FIC 3型由编码多药耐药蛋白3(MDR3)的ABCB4基因突变所致。MDR3是位于毛细胆管的磷脂转位酶,负责将磷脂酰胆碱从肝细胞内转至毛细胆管中。与其他的FIC不同,FIC 3型患者的GGT通常明显升高,组织学检查除发现门管区炎症和纤维化/肝硬化外,尚有弥漫性胆管增生。FIC 3型可伴发肝内胆石症。近期又有报道[30]在脑信号蛋白7A基因纯合子R148W突变引起PFIC。
目前尚无治疗FIC药物,常用治疗方法有UDCA和胆汁酸肠肝循环阻断剂,另外普遍推荐予以补充中链甘油三酯和脂溶性维生素。对于FIC 3型,UDCA可改善生化指标,使部分轻型患者长期无病生存。利福平可缓解瘙痒。对晚期FIC建议肝移植治疗。
推荐意见10:家族性肝内胆汁淤积症(FIC)是一组常染色体隐性遗传疾病,以瘙痒和黄疸为主要表现,可表现为轻重不等的谱系。(B1) 基因检测是确诊的金标准。(B1) FIC 1、2、4、5和6型以低GGT、严重瘙痒为特征,可伴有不同的肝外表现。FIC3型以高GGT为特征。FIC尚无有效的治疗方法。(C2) UDCA可改善部分FIC3型患者肝功能指标。(C2) 胆汁分流对部分FIC患者肝生化指标有益。(C2) 晚期患者推荐肝移植评估。(B1)
8.3 Alagille综合征 由Notch信号途径中的JAG1或NOTCH2基因突变引起常染色体显性遗传,约94%由JAG1基因突变引起,2.5%由NOTCH2基因突变引起。该病以儿童及青少年高GGT和不同程度的肝外脏器(心血管系统、骨骼、肾脏、眼睛和颜面等)受累为特征,估计发病率1/30 000至1/70 000之间,基因型和表型无明显关系[31-32]。肝活检病理发现小叶间胆管减少或缺乏曾被认为是该病的最重要特征,但少部分患者可无小叶间胆管减少或缺乏,甚至在疾病早期可有小胆管增生。诊断标准为符合下列之一:(1)肝组织学检查发现小叶间胆管减少或缺乏时,满足主要临床特征(包括慢性胆汁淤积、心脏杂音、蝴蝶椎骨、眼睛异常、肾脏异常和特征性的面容)中至少3项;(2)无肝组织学小叶间胆管减少或缺乏证据时,满足主要临床特征(见上)至少4项;(3)有明确该病家族史,或发现基因突变,满足至少2项主要临床特征。Alagille综合征缺乏满意的治疗方法,主要是对症处理(UDCA、阻断胆汁酸肠肝循环药物)和脂溶性维生素补充。近期美国食品药品监督管理局已批准LIRVMARI(Maralixibat)口服溶液用于治疗1岁及以上的Alagille综合征患者的胆汁淤积性瘙痒,LIRVMARI是一种回肠胆汁酸转运蛋白(IBAT)抑制剂,阻断胆汁酸肠肝循环[33]。
推荐意见11:Alagille综合征主要发生于儿童及青少年,由JAG1或NOTCH2基因突变导致小叶间胆管减少,从而引起胆汁淤积伴瘙痒,及心血管系统、眼、骨骼、面部异常为特征多系统损害,以对症和支持治疗为主。(C2)
9 ICP
ICP的发病机制涉及多种因素,遗传、激素和环境因素起重要作用。患ICP后经母体流向胎儿的胆汁酸增加,表现为胆汁酸在羊水、脐带血和胎粪中升高。孪生妊娠时ICP发病率升高以及大剂量服用避孕药和孕酮可诱发ICP支持激素在该病发生中起关键作用[34-35]。同一家族成员ICP发病率增加以及种族间的差异说明了遗传因素的作用。最近遗传学研究鉴定出毛细胆管转运蛋白基因变异体(ABCB4、ABCB11、ABCC2和ATP8B1)和某些ICP患者调节物(FXR)。妊娠过程中,当激素和其他底物超过毛细胆管转运体转运能力时,毛细胆管转运体轻度功能不全就能诱发胆汁淤积。因此如产后胆汁淤积(伴有GGT水平升高)持续存在,可考虑进行ABCB4等突变分析。
9.1 诊断 胆汁淤积出现于妊娠晚期,在分娩后自发地迅速改善。特征为:(1)妊娠期严重瘙痒(多数起始于妊娠第2或第3阶段);(2)ALT和空腹胆汁酸和甘胆酸升高;(3)产后(4~6周内)症状和体征自发缓解[34-36]。瘙痒导致孕妇不适和烦恼,ICP也增加早产和胎儿猝死风险。一般预后较好,但黄疸久者,因缺乏维生素K依赖性凝血因子Ⅱ、Ⅶ、Ⅹ,产时可发生大出血。本症对胎儿影响较大,发生胎儿宫内窘迫、早产和死胎的危险性相对较高。ICP可根据临床表现结合甘胆酸及总胆汁酸(TBA)2个指标,血甘胆酸升高≥10.75 μmol/L或TBA升高≥10 μmol/L诊断,产后肝生化指标完全正常后可确诊。ABCB4变异的ICP患者GGT水平升高,否则GGT正常。10%~15%患者血清结合胆红素中度升高出现轻度黄疸。ICP诊断要求排除其他疾病,且通过产后访视复查做出最后诊断。肝活组织检查通常是非必需的。
妊娠期肝生化指标异常除ICP外,尚需考虑先兆子痫、HELLP综合征和妊娠急性脂肪肝[4],这3种疾病有相当多的重叠。先兆子痫是妊娠期血压升高的基础上出现脏器损害,脏器损害是多方面的,比较常见的是肾脏损害,孕妇表现为尿蛋白阳性。还会造成肝生化指标异常、血小板下降和溶血等。HELLP综合征以溶血、肝酶升高和血小板减少(<50×109/L)为特点,是妊娠期高血压疾病的严重并发症,多数发生在产前,胆红素<85 μmol/L,影像学显示肝坏死、血管瘤、肝破裂。妊娠急性脂肪肝是一种妊娠晚期的急性肝脂肪变,大多累及年轻初产妇,多在妊娠最后3个月内或产后早期发生,起病急骤,预后凶险,临床表现如同急性重型肝炎,表现为急性肝功能衰竭,常伴有肾功能衰竭,影像学显示肝脂肪变。
9.2 治疗 UDCA可作为治疗ICP一线药物,可改善67%~80%ICP患者瘙痒和肝生化指标[34-36]。SAMe疗效逊于UDCA,但有附加效果。如果经数天UDCA标准治疗后瘙痒无适当减轻,可选择SAMe或考虑利福平。药物治疗同时还注意产科情况。强调治疗过程中加强胎儿监护,把握终止妊娠时机,对降低围生儿死亡率具有重要意义。妊娠35周后,若出现病情进展、宫缩不能抑制、胎动异常、胎心率变异或应激试验无反应、羊水胎粪污染等,应把握时机,积极终止妊娠[4]。
推荐意见12:ICP诊断依据:(1)妊娠期瘙痒;(2)血清ALT水平以及空腹胆汁酸和甘胆酸水平升高;(3)除外其他原因的肝功能异常或瘙痒。产后肝生化指标完全正常后可确诊。(B2)
推荐意见13:UDCA和S-腺苷蛋氨酸可用于妊娠第2或第3期有胆汁淤积且有症状的患者,可缓解瘙痒并能改善肝生化指标(B1),但尚无胎儿保护和减少并发症的方法。(C2)
10 肝外表现及处理
10.1 瘙痒 瘙痒是一种仅有皮肤不快感觉而无原发性皮肤损害的症状,这种感觉无论在性质、持续性及定位均不同于触觉和痛觉。瘙痒的存在本身无预后价值,并不反映疾病内在的严重程度。瘙痒的发病机制尚不清楚,大多数认为与血清自分泌运动因子(autotaxin,ATX)活性增加和溶血磷脂酸形成有关。此外,胆汁酸盐、内源性阿片肽、5-羟色胺(5-HT)、感觉神经元的过度兴奋、雌激素和孕激素、肝肠瘙痒原改变、遗传因素等也相关[37-38]。瘙痒与胆汁淤积的关系说明引起瘙痒的物质是在胆汁中正常排泄的某些物质。肝细胞衰竭时瘙痒消失表明引起瘙痒的物质是由肝细胞产生,而血清胆酸依然很高[4]。瘙痒的严重度评分有下列3种[38-39]:(1)视觉模拟评分(visual analogue scale,VAS):瘙痒严重性按皮肤抓痕分为:抓痕、斑块、结节和/或疤痕,根据轻中重程度分别评分0~3(4)级,总分从0分无瘙痒到10分严重瘙痒。(2)瘙痒严重程度量表(itch severity scale,ISS)包括频率、睡眠、心情、性欲、性功能、李克特量表(Likert scale)评估瘙痒强度、瘙痒涉及的体表面积7项,总评分范围可以从0分无瘙痒到21分即最严重的瘙痒。(3)半定量评估瘙痒:根据瘙痒频率分为4个阶段:偶尔瘙痒、无临床症状的每天间断性瘙痒、出现临床症状的每天间断性瘙痒和持续性瘙痒。多个药物可以单独或者联合应用治疗瘙痒,包括考来烯胺、抗组胺药、孕烷X受体激动剂、阿片受体拮抗剂、5-HT受体拮抗剂等。考来烯胺作为胆汁淤积性瘙痒的一线治疗药物[5,40]。治疗瘙痒推荐剂量是4 g/d,最大不超过16 g。注意应与其他药物(尤其是UDCA)间隔4~6 h服用以避免影响其他药物的吸收。UDCA也可采用顿服的给药方式,避免消胆胺对药物吸收的影响。孕烷X受体激动剂利福平可改变瘙痒原的代谢,下调ATX减少溶血磷脂酸的形成,减轻瘙痒症状而作为二线治疗药物,尤其是考来烯胺不耐受或治疗效果不著的患者[41]。利福平开始一般以150 mg/d单剂口服,有效后继续服用。如无效,可隔周阶梯增加剂量至600 mg/d。利福平治疗美沙酮(一种镇痛药)成瘾患者时可引起阿片戒断反应,因此利福平可能存在阿片拮抗作用,进而缓解胆汁淤积性瘙痒。利福平应用后患者尿色变红,还可出现中毒性肾损害、肝毒性,偶有溶血发生。由于利福平有潜在的肝损害,用药期间必须密切监测肝生化指标。口服阿片受体拮抗剂纳曲酮25~50 mg/d作为三线治疗瘙痒的药物,少数患者可有恶心、呕吐、轻度疼痛等副作用。纳曲酮的代谢产物可以在失代偿性肝病患者体内积聚,因此这些患者使用此药时需谨慎。这类药物应先小剂量使用,再逐渐提高剂量,以免引起类似麻醉药的戒断作用。如上述药物均无效也可应用选择性5-HT再摄取抑制剂舍曲林治疗,作为四线治疗药物,初始剂量1次50 mg/d,数周后增加至100 mg/d[40]。近年来,紫外照射、体外白蛋白透析和鼻胆管引流也用于改善胆汁淤积性瘙痒,并获得较好的疗效。应用药物和其他方法疗效不佳的顽固性瘙痒患者建议进行肝移植评估。
推荐意见14:考来烯胺是治疗瘙痒一线药物,推荐剂量是4 g/d,最大剂量不超过16 g/d。注意应与其他药物(尤其是UDCA胆酸)间隔4~6 h服用以避免影响其他药物的吸收。(B2)
推荐意见15:利福平为瘙痒治疗二线药物,开始一般以150 mg/d单剂口服,有效后继续服用。如无效,可隔周阶梯增加剂量至300 mg/d。利福平有潜在的肝损害,用药期间必须密切监测肝生化指标。(C2)
推荐意见16:口服阿片受体拮抗剂纳曲酮为瘙痒治疗三线药物,先小剂量口服25 mg/d,无效后再逐渐提高剂量至50 mg/d,以免引起类似麻醉药的戒断作用。(C1)
推荐意见17:选择性5-HT再摄取抑制剂舍曲林为瘙痒治疗四线药物,初始剂量50 mg/d,数周后可增加至100 mg/d。(C2)
推荐意见18:瘙痒经上述药物治疗无效者可考虑选用紫外线照射、体外白蛋白透析及鼻胆管引流等方法。(C2) 药物和其他方法疗效不佳的严重瘙痒患者考虑肝移植。(C2)
10.2 疲劳 胆汁淤积患者常常有疲劳症状,尤其是PBC,可出现于70%~80%的慢性胆汁淤积患者[4]。疲劳是一个复杂的症状,包括持续的衰竭感觉,正常工作能力缺失,心理和生理功能的下降。由于其是非特异性症状,目前常用疲劳影响评分(fatigue impact score,FIS)或PBC-40等方法评估,目前对疲劳的发病机制依旧不清楚,也无有效的治疗方法[42-43]。治疗前需排除患者的贫血、糖尿病、甲状腺功能减退、肾和肾上腺功能不全及抑郁等表现。目前可能的治疗方法和药物有选择性5-HT3受体拮抗剂如昂丹司琼、阿片受体拮抗剂和中枢神经兴奋药莫达非尼等治疗,莫达非尼起始剂量100 mg,根据患者耐受情况和对药物的反应逐渐增加剂量到200 mg,但其疗效有待进一步研究证实。UDCA是PBC治疗的有效药物,对伴随的疲劳症状无明显的改善作用。肝移植也不能降低疲劳的发生,甚至在肝移植1年后,疲劳仍是患者痛苦的症状,尽管疲劳程度减轻。提倡健康的生活方式,包括保证足够的睡眠、有规律的锻炼、戒酒和晚上戒咖啡等都有好处。抗抑郁药可以部分减轻抑郁患者的疲劳。
10.3 黄色瘤 常见于慢性胆汁淤积患者,扁平或略高出皮肤表面、黄色、柔软,通常见于眼周,也可见于掌褶、乳房下和颈部、胸部和后背。其发生与血脂水平相关,通常胆固醇需超过4.5 g/L以上才会出现。在黄疸消退或晚期肝细胞衰竭时,如果胆固醇水平下降,这些表现可消失。黄色瘤不需要特殊处理。
10.4 脂代谢紊乱 胆汁淤积患者由于胆汁酸升高而抑制胆固醇的代谢,常存在脂质紊乱,患者胆固醇和甘油三酯均可升高;目前尚无证据表明它可增加动脉粥样硬化的危险性。通常不需要降脂治疗,他汀和贝特类药物可安全用于胆汁淤积性肝病患者伴有脂代谢紊乱的患者[44],考来烯胺有改善血脂异常作用。
10.5 脂肪泻 胆汁淤积后肠道内缺乏足够的胆盐,导致脂肪性食物及脂溶性维生素(A、D、E和K)消化吸收障碍。脂肪泻与黄疸深度成正比,表现为大便量大、松软、油质过多,便灰白,有异味。
推荐意见19:胆汁淤积性疲劳需排除患者贫血、糖尿病、甲状腺功能减退、肾和肾上腺功能不全及抑郁等表现。建议保证足够睡眠、规律锻炼、戒酒和晚上戒咖啡。(C2) 选择性5-HT3受体拮抗剂如昂丹司琼、阿片受体拮抗剂和中枢神经兴奋药莫达非尼(100~200 mg/d)等有一定疗效。(C2) 抗抑郁药可以部分减轻抑郁患者的疲劳。(C2) 肝移植对疲劳改善无明显作用。(C2)
推荐意见20:黄色瘤不需要特殊处理。(B2) 他汀和贝特类药物可用于伴有脂代谢紊乱的患者,考来烯胺有改善血脂异常作用。(B2)
10.6 肝性骨营养不良 骨骼疾病是慢性肝脏疾病的并发症,可发生骨骼疼痛和骨折,发生机制为骨质疏松和骨软化。骨质疏松存在骨基质和矿物质丢失,骨软化存在类骨质的矿化缺陷。骨质疏松诊断基于应用双能X线吸收测量仪(dual energy X-ray absorptiometry,DEXA)测定骨矿物质密度(bone mineral density,BMD)[4-5,45]。参照世界卫生组织推荐的诊断标准,DEXA测定骨密度值低于同性别、同种族健康成人的骨峰值不足1个标准差属正常(T值≥-1.0 SD);降低l~2.5个标准差为骨量低下或骨量减少(-2.5 SD 10.7 脂溶性维生素缺乏 胆汁淤积时肝脏分泌胆汁到小肠障碍,肠内胆盐减少,可出现脂溶性维生素缺乏和脂肪泻,因此需要适当补充脂溶性维生素[45]。如凝血酶原时间延长,肌注维生素K(10 mg/d),直至正常。因维生素A所致的夜盲,口服维生素A 25 000~50 000 IU/d。维生素E缺乏少见,有报道在儿童可见,表现为小脑共济失调、后索功能障碍、末梢神经病变和视网膜变性,可口服予以补充。建议测定血液脂溶性维生素的水平以指导其补充的需要,但目前尚没有得到普遍使用和推广。 推荐意见21:建议患者补充钙及维生素D预防骨质疏松。成人每日钙摄入量800 mg;绝经后妇女和老年人每日钙摄入推荐量为1000 mg。维生素D的成年人推荐剂量为200 IU/d;老年人推荐剂量为400~800 IU/d。(C1) 二膦酸盐类(如阿仑膦酸盐70 mg/周或伊班膦酸盐150 mg/月或其他同类药物)可治疗和预防骨质疏松。(C2) 每年骨密度测量可用于骨质疏松治疗和随访。(C2) 推荐意见22:注意脂溶性维生素的监测和补充。凝血酶原时间延长,注射维生素K1 10 mg/d。(B1) 维生素A所致的夜盲,口服维生素A 25 000~50 000 IU/d。(C1) 维生素E缺乏少见,可口服予以补充10~100 mg/d。(C2) 尽管近年来在胆汁淤积性肝病的诊断和治疗方面有不少进展,但该领域仍面临诸多问题和挑战。在基础研究方面,有关胆汁淤积的发生机制尤其是其分子机制、胆汁酸转运蛋白遗传和变异对胆汁淤积发生和发展的影响、胆汁淤积中胆汁酸成分对肝脏及全身的影响如何有待进一步研究;PBC和PSC是最重要的胆汁淤积性肝病,其病因尚未阐明,UDCA是PBC的主要药物,但对应答不佳的患者治疗仍有不少未满足的需求,PSC尚未有有效的治疗药物。胆汁淤积性肝病的流行病学、诊断标志物和诊断标准尚需进一步完善和验证;治疗上尚需要更有效的药物和方法等。 执笔人:陆伦根、蔡晓波、王建设、曲颖、尤红、马雄、韩英、南月敏、徐小元、段钟平、魏来、贾继东、庄辉 指南制定专家(以姓氏拼音排序):蔡晓波(上海交通大学附属第一人民医院消化内科)、陈莎(首都医科大学附属北京友谊医院肝病中心)、陈煜(首都医科大学附属北京佑安医院肝病中心)、陈红松(北京大学人民医院肝病研究所)、崔丽娜(空军军医大学第一附属医院消化内科)、董加强(空军军医大学第一附属医院消化内科)、窦晓光(中国医科大学附属盛京医院感染科)、段维佳(首都医科大学附属北京友谊医院肝病中心)、段钟平(首都医科大学附属佑安医院肝病中心)、郭长存(空军军医大学第一附属医院消化内科)、郭冠亚(空军军医大学第一附属医院消化内科)、韩涛(天津市第三中心医院肝内科)、韩英(空军军医大学第一附属医院消化内科)、侯金林(南方医科大学南方医院感染内科)、胡鹏(重庆医科大学附属第二医院感染科)、宦怡(空军军医大学第一附属医院放射影像科)、贾继东(首都医科大学附属北京友谊医院肝病中心)、孔媛媛(首都医科大学附属北京友谊医院国家消化系统疾病临床医学研究中心方法学平台)、李杰(北京大学医学部基础医学院病原生物学系)、李军(江苏省人民医院感染病科)、李淑香(首都医科大学附属北京友谊医院肝病中心)、李增山(空军军医大学第一附属医院病理科)、令狐恩强(解放军总医院第一医学中心消化内科)、刘家云(空军军医大学第一附属医院检验科)、刘景丰(福建医科大学孟超肝胆医院肝胆外科)、刘燕敏(首都医科大学附属北京佑安医院肝病科)、刘迎娣(解放军总医院第一医学中心消化内科)、陆伦根(上海交通大学附属第一人民医院消化内科)、罗新华(贵州省人民医院感染科)、吕婷婷(首都医科大学附属北京友谊医院肝病中心)、马雄(上海交通大学医学院附属仁济医院消化内科)、苗琪(上海交通大学医学院附属仁济医院消化内科)、南月敏(河北医科大学第三医院中西医结合肝病科)、曲颖(上海交通大学附属第一人民医院消化内科)、任红(重庆医科大学第二医院传染科)、任万华(山东省立医院感染性疾病科)、尚佳(河南省人民医院感染性疾病科)、尚玉龙(空军军医大学第一附属医院消化内科)、时永全(空军军医大学第一附属医院消化内科)、唐承薇(四川大学华西医院消化内科)、王建设(复旦大学附属儿科医院感染科)、王婧雯(空军军医大学第一附属医院药剂科)、王绮夏(上海交通大学医学院附属仁济医院消化内科)、魏来(北京大学人民医院肝病科)、吴浩(四川大学华西医院消化内科)、徐小元(北京大学第一医院感染疾病科)、阎明(山东大学齐鲁医院消化内科)、杨东亮(华中科技大学同济医学院附属协和医院感染科)、杨永峰(南京市第二医院肝病科)、杨诏旭(空军军医大学第一附属医院肝胆外科)、尤红(首都医科大学附属北京友谊医院肝病中心)、张欣欣(上海交通大学医学院附属瑞金医院感染科)、张跃新(新疆医科大学第一附属医院感染科)、赵景民(解放军总医院第五医学中心病理科)、赵守松(蚌埠医学院第一附属医院感染病科)、赵新颜(首都医科大学附属北京友谊医院肝病中心)、郑林华(空军军医大学第一附属医院消化内科)、周新民(空军军医大学第一附属医院消化内科)、庄辉(北京大学医学部基础医学院病原生物学系) 志谢(以姓氏拼音排序):安纪红(内蒙古自治区人民医院感染科)、邓国宏(陆军军医大学第一附属医院传染科)、黄燕(中南大学湘雅医院传染科)、黄缘(北京清华长庚医院肝胆内科)、李荣宽(大连医科大学附属二院感染科)、李树臣(哈尔滨医科大学第二附属医院传染科)、陆海英(北京大学第一医院传染科)、石荔(西藏自治区人民医院感染科)、苏明华(广西医科大学第一附属医院感染科)、温志立(南昌大学第二附属医院消化内科)、吴彪(海南省人民医院感染科)、徐京杭(北京大学第一医院肝病科)、杨丽(四川大学华西医院消化内科)、杨积明(天津市第二人民医院感染科)、杨晋辉(昆明医科大学附属二院消化内科)、张缭云(山西医科大学第一医院传染科)、周璐(天津医科大学总医院消化内科)、周永健(广州市第一人民医院消化内科)、祖红梅(青海省第四人民医院传染科)参加本指南制定的讨论,并提出了富有建设性的意见和建议。 利益冲突声明:所有作者均声明不存在利益冲突。 参考文献见二维码11 待解决的问题

猜你喜欢
基层中医药(2022年7期)2022-11-17
珠江水运(2022年17期)2022-09-25
传染病信息(2022年2期)2022-07-15
中国典型病例大全(2022年12期)2022-05-13
智慧医学(2021年1期)2021-09-10
珠江水运(2021年15期)2021-08-29
家庭医学·下半月(2021年1期)2021-03-28
家庭医学(2018年2期)2018-07-14
黄河黄土黄种人(2017年11期)2017-11-27
水能经济(2017年6期)2017-10-19
