
蒸煮对小米膳食纤维结构及功能特性的影响
2022-02-08 04:03:26徐晓琴陈金凤尤莺鸽陈雪琴李志程张晓萌张盛贵
中国粮油学报 2022年11期
徐晓琴,陈金凤,尤莺鸽,陈雪琴,李志程,张晓萌,张盛贵
(甘肃农业大学理学院1,兰州 730070) (甘肃农业大学食品科学与工程学院2,兰州 730070)
膳食纤维(DF)可以增加排便量、刺激胃肠运动、降低血糖和胆固醇水平等[1],增加DF摄入量可以降低糖尿病、肥胖和心血管等慢性疾病的风险[2]。根据水中溶解度不同,DF可分为不溶性膳食纤维(IDP)和可溶性膳食纤维(SDF),IDF和SDF在不同的原料中含量不同,并具有不同的性质[3]。DF主要来自谷物、水果、坚果和蔬菜。小米膳食纤维主要存在于种皮及少量的糊粉层中[4]。在天然植物细胞壁中,细胞壁的结构骨架是纤维素,半纤维素和木质素分散在纤维中,导致DF产率低、功能性差[5]。因此,通过合适的改性方法提高DF的提取率和功能特性,从而提高人体对DF的利用率,已经成为食品加工领域的研究热点。
小米作为甘肃的主要杂粮作物,具有耐储藏、耐旱、适应性强等优点,其营养均衡、消化率高,具有抗氧化、消炎、抗癌、抗菌等生理作用[9]。因此,本研究以“陇谷032”小米为原料,首先通过蒸煮处理对小米进行改性,再经过酶法处理提取DF,探讨蒸煮处理对DF结构和功能特性的影响,以期为甘肃小米DF的开发和利用提供参考。
1 材料与方法
1.1 材料与设备
1.1.1 实验材料
“陇谷032”小米、蛋白酶(1 000 U/g,分析纯)、糖化酶(100 000 U/mL,分析纯)、高温α-淀粉酶(20 000 U/mL,分析纯)。
1.1.2 仪器与设备
X’Pert-Pro MPD多晶粉末X射线衍射仪,TGA50热重分析仪和DSC25差示扫描量热仪,JSM-6701F冷场发射型扫描电子显微镜,NEXUS670傅里叶红外光谱仪,Bettersize 2600激光粒度分析仪。
1.2 方法
1.2.1 蒸煮小米制备
小米室温浸泡3 h(1∶1,g/mL),蒸锅中平摊成2 cm薄层,常压(98 ℃)蒸制1 h(水沸腾时计时),手捻易碎且有完整颗粒时取出放入30 ℃烘箱中干燥12 h后磨粉,过40目筛。装入密封袋储存于4 ℃冰箱备用。以未蒸煮的小米作为对照。
1.2.2 小米膳食纤维提取
参考AOAC法和Liu等[10]的方法。小米粉用正己烷(1∶2.5,g/mL)脱脂,称取10.0 g干燥的小米粉,加入250 mL的磷酸盐缓冲液(pH 6),500 μL高温α-淀粉酶,95 ℃酶解30 min。冷却后将pH调为7.5,加入1 mL碱性蛋白酶(5 mg/mL),60 ℃水浴30 min。最后将pH调为4.5,加入3 mL糖化酶,60 ℃水浴30 min后离心(5 000 r/min,10 min)分离沉淀和上清液。沉淀依次用蒸馏水、95%乙醇和丙酮清洗3次后再次离心,所得的沉淀即为IDF。收集离心后的上清液,加入60 ℃,95%的乙醇(上清液∶乙醇的体积比1∶4)混匀后静置12 h,得到的沉淀即为SDF。IDF和SDF经真空冷冻干燥后磨粉,过100目筛。
1.2.3 小米膳食纤维成分测定
在干质量的基础上,参照GB/T 5009.3—2016《食品安全国家标准 食品中水分的测定》、淀粉测定参照淀粉含量检测试剂盒、GB/T 5009.4—2016《食品安全国家标准 食品中灰分的测定》、GB/T 5009.5—2016《食品国家安全标准 食品中蛋白质的测定》、GB/T 5009.6—2016《食品安全国家标准 食品中脂肪的测定》分别测定提取的膳食纤维的水分、灰分、粗蛋白、脂肪含量测定。
1.2.4 小米膳食纤维结构性质测定
1.2.4.1 扫描电镜测定
利用扫描电镜对样品结构进行表征,用双面导电胶带将样品固定在样品台上,在1 000倍下观察样品颗粒形貌。
1.2.4.2 傅里叶红外光谱测定
样品与KBr粉末(1∶100)混匀研磨后压片,在400~4 000 cm-1波数下扫描,分辨率为4 cm-1。
1.2.4.3 X射线衍射的测定
用X射线衍射仪对样品的晶体结构进行分析,扫描速度为1(°)/min,衍射角(2θ)范围为10°~80°。
1.2.4.4 粒径测定
用激光粒度分析仪对样品的粒径进行分析。以蒸馏水作为分散剂,通过超声辅助使样品分散均匀,设定折射率1.460,粒径测定范围0.01~10 000 μm。
1.2.4.5 热稳定性测定
参考李璐等[11]的方法,利用TGA和DSC对样品热特性进行分析,TGA测定参数:升温范围在25~800 ℃之间,升温速率为10 ℃/min,N2流速35 mL/min。DSC测定参数:升温范围20~200 ℃,升温速率10 ℃/min,N2流速50 mL/min。
1.2.5 小米膳食纤维功能性质测定
1.2.5.1 SC、WHC、OHC测定
参考Ma等[12]的方法,称取0.5 g样品于10 mL量筒中,加入5 mL蒸馏水,室温放置24 h,记录体积;用式(1)计算SC。
SC=(V1-V0)/W0
(1)
式中:V0为膨胀前的体积/mL;V1为膨胀后的体积/mL;W0为样品质量/g。
参考Ma等[12]的方法,称取0.5 g样品于已知质量的离心管中,加入20 mL蒸馏水混匀,室温振荡12 h后,5 000 r/min离心10 min。弃去上清液,吸干离心管管壁残留的水分,称离心管和沉淀的总质量。用式(2)计算WHC。
WHC=m1/m0
(2)
式中:m1为离心后样品质量/g;m0为样品质量/g。
参考Ma等[12]的方法,称取0.5 g样品于已知质量的离心管中,加入20 mL菜籽油混匀,室温振荡12 h后,5 000 r/min离心10 min,弃去上清液,吸干离心管管壁残留的菜籽油,用式(3)计算OHC。
OHC=m1/m0
(3)
式中:m1为离心后样品质量/g;m0为样品质量/g。
1.2.5.2 胆固醇吸附能力测定
参考Ma等[12]的方法,胆固醇用新鲜蛋黄代替,蛋黄与蒸馏水(体积比1∶9)充分搅拌呈乳液状态。取0.5 g样品加入15 mL的蛋黄乳液,调节pH至2.0(模拟胃)和7.0(模拟小肠),37 ℃振荡2 h,4 000 r/min离心20 min。用式(4)计算胆固醇吸附量。
胆固醇吸附量=(n1-n2)/m1
(4)
式中:n1为吸附前胆固醇的量/mg;n2为吸附后胆固醇的量/mg;m1为样品质量/g。
1.2.5.3 亚硝酸盐吸附能力测定
参考Luo等[13]的方法,取0.5 g样品加入50 mL 100 μmol/L亚硝酸钠溶液,调节pH至2.0(模拟胃)和7.0(模拟小肠),37 ℃振荡2 h,吸取2 mL上清液,测定吸光度,用式(5)计算亚硝酸盐吸附量。
亚硝酸盐吸附量=(n1-n2)/m1
(5)
式中:n1为吸附前亚硝酸盐的量/μg;n2为吸附后亚硝酸盐的量/μg;m1为样品质量/g。
1.3 数据统计与分析
所有实验平行测定3次。数据采用Excel 2010软件计算平均值和标准差,通过SPSS 22.0软件中t检验进行显著性分析,P<0.05表示差异显著,采用Origin 8.0软件作图。
2 结果与分析
2.1 小米膳食纤维成分分析
脱脂后的小米经酶处理制备小米膳食纤维,得到的小米膳食纤维中水分、淀粉、灰分和蛋白质的质量分数分别为2.07%、0.12%、0.08%和0.03%,未检测出脂肪。
2.2 小米膳食纤维结构性质分析
2.2.1 扫描电镜分析
由图1观察可知,4种样品结构存在明显差异,IDF和C-IDF表面呈明显片状结构,碎片不规则,存有孔洞,C-IDF孔洞更多,表面褶皱明显,结构更为松散;这与Wang等[14]通过酶法提取猕猴桃膳食纤维,观察到不溶性膳食纤维孔较多,且结构更加松散、复杂的结果相似。SDF和C-SDF均呈现不规则的块状结构,表面有小团块,C-SDF的表面出现较大的团聚体,相互堆积形成紧密相连的组织;这与Liu等[15]通过酶处理麦麸可溶性膳食纤维的结果类似。C-IDF和C-SDF结构存在差异,可能与蒸煮处理后IDF和SDF降解和聚集有关。蒸煮导致IDF中木质素、纤维素和半纤维素裂解成小分子物质[16],使得C-IDF表面变得疏松、粗糙,微观结构发生变化。此外,蒸煮处理增强了颗粒间的结合,使DF结构更有黏结性[17],从而导致C-IDF和C-SDF颗粒形态的改变。由此可知,IDF和SDF微观结构存在差异,蒸煮能改变DF的微观结构。扫描电镜结果为DF与胆固醇、亚硝酸盐的结合提供了微观结构基础。

图1 IDF、C-IDF、SDF和C-SDF的扫描电镜图(1 000倍)
2.2.2 傅里叶红外光谱分析
如图2所示,IDF、C-IDF、SDF和C-SDF在3 420 cm-1附近的宽吸收峰是O—H的伸缩振动,IDF和C-IDF的吸收峰主要是由木质素、纤维素和半纤维素分子内或分子间的O—H伸缩振动,SDF和C-SDF的吸收峰主要来自甘露糖、半乳糖和阿拉伯糖中的—OH基团;C-IDF在此处的峰面积变宽,强度增加,说明蒸煮处理使得纤维素、半纤维素中部分糖苷键断裂,氢键增强,亲水性增强[16],符合C-IDF具有较高WHC的结果。2 927cm-1处的吸收峰是由糖类甲基和亚甲基上C—H的伸缩振动引起的,2 360 cm-1处的吸收峰是C—H的反伸缩振动,C-SDF在这两处的峰强度增加,表明C-SDF中存在典型的糖类化合物。IDF、C-IDF、SDF和C-SDF在1 639 cm-1处的吸收峰主要来自—COOH伸缩振动,表明样品中含有糖醛酸,这与Huang等[18]通过高温蒸煮辅助酶法改性茶渣膳食纤维中的研究结果一致。另外,4种样品在920 cm-1附近有β-糖苷键吸收峰,在860 cm-1附近有α-糖苷键吸收峰[17]。除此之外,IDF和C—IDF在900~1 200 cm-1之间的吸收峰是由半纤维素的酰基氧(CO—OR)伸缩振动和木质素酰基的C—O伸缩振动引起的[19]。SDF和C—SDF在1 000~1 200 cm-1区域内的强吸收峰是由C—O—C糖苷键,C—C和C—OH键伸缩振动引起的。C-SDF的吸收峰强度降低,可能是由于蒸煮处理使样品中部分水溶性多糖发生降解。SDF和C—SDF在1 745~1 750 cm-1范围内无振动信号峰,说明SDF和C-SDF中的果胶为低酯果胶[20],低酯果胶具有良好的安全性和生物降解性,与金属离子发生反应,形成网状凝胶,有效去除重金属离子[21]。IDF、C-IDF、SDF和C-SDF的红外光谱相似,说明蒸煮没有改变小米DF的主要官能团(如—OH、—COOH、—CHO等)。但其含量或结合程度不同,导致某些吸收峰强度不同。
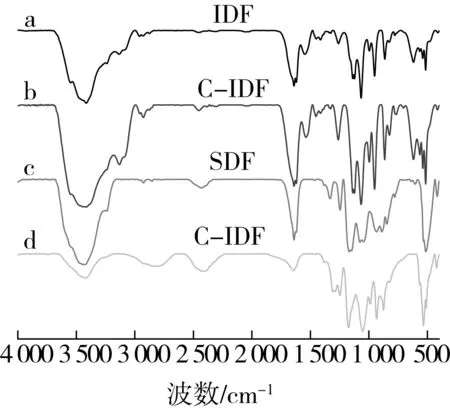
图2 IDF、C-IDF、SDF和C-SDF的红外光谱图
2.2.3 X射线衍射分析
IDF、C-IDF与SDF、C-SDF的组分、分子量不同,使其结晶衍射峰不同(图3)。IDF在扫描角度2θ为20.02°处表现出强烈的结晶衍射峰,C-IDF结晶区被破坏,结晶强度减弱,衍射角变宽,可能因为蒸煮处理后木质素、纤维素、半纤维素之间发生了相互转化[22]。SDF在2θ为29.20°处有明显的结晶衍射峰,在16.30°、49.48°、56.33°处有较弱的衍射峰,而C-SDF仅在23.95°和28.43°处有明显的特征峰。表明C-SDF的部分结晶区被破坏,晶体结构由有序向无序转变。SDF主要是由半乳糖醛酸、阿拉伯糖、半乳糖等成分组成,在蒸煮过程中多糖和低聚糖在一定程度上被分解为单糖,使得C-SDF结晶区发生变化[17]。C-IDF和C-SDF分子之间的相互作用力变弱,组织变得疏松,不仅有助于降低DF的聚合度,还能有效提高其溶解性[23],从而使其SC、WHC和OHC增强,进而改善DF的生理功能。


图3 IDF、C-IDF、SDF和C-SDF的X射线衍射图
2.2.4 粒径分析
与IDF相比,C-IDF、SDF和C-SDF粒径分布峰均发生左移,SDF和C-SDF粒径分布峰高于IDF和C-IDF,峰跨度窄,表明SDF和C-SDF粒度分布相对集中,颗粒尺寸均匀。IDF粒径分布峰较宽,有2个峰出现,说明其颗粒尺寸大小不一;C-IDF粒径分布峰宽度变小,峰值升高,仅出现1个峰,表明蒸煮处理后颗粒大小越来越均匀(图4)。IDF、C-IDF、SDF和C-SDF的体积平均粒径大小依次为IDF >C-IDF > C-SDF > SDF(表1)。IDF的D(4,3)显著高于C-IDF的D(4,3)(P<0.05),蒸煮处理使得部分DF被破坏,质地变得松散[10],从而使C-IDF颗粒粒径减小;与SDF相比,C-SDF的D(4,3)增加,由扫描电镜结果可印证,处理后C-SDF颗粒发生部分团聚,从而使粒径增加。

图4 IDF、C-IDF、SDF和C-SDF的粒径分布图

表1 IDF、C-IDF、SDF和C-SDF的粒径参数
2.2.5 热稳定性分析
如图5所示,由于IDF和SDF组成成分不同,故IDF、C-IDF与SDF、C-SDF的质量损失及质量损失速率差异较大。IDF和C-IDF在200 ℃之前,主要是样品内游离水及结合水的挥发,质量损失缓慢;从200 ℃开始是第二阶段热分解反应,此阶段内IDF和C-IDF质量损失最多,质量损失分别为79%和65%。随温度的升高,IDF和C-IDF的质量损失速率分别在320 ℃和300 ℃左右达到最大,之后质量损失速率减小,IDF的温度达到560 ℃后,其质量损失速率基本保持不变,而C-IDF的质量损失速率一直减小。蒸煮处理后,纤维素、半纤维素、木质素之间发生解聚反应[22],使部分IDF转化成SDF,使得C-IDF质量损失降低。SDF和C-SDF在420 ℃之前,一直处于TGA损失阶段,质量损失分别为25%和24%,此阶段主要涉及样品内自由水、结合水的挥发,以及半乳聚糖、阿拉伯聚糖、阿拉伯半乳聚糖等的分解。图5b中,SDF和C-SDF在70、190 ℃时出现2个质量损失速率峰,且C-SDF的峰值更高,可能是SDF和C-SDF的表面出现团聚体,结构变得紧密,水分不易从分子内蒸发,第2个峰的出现是因为SDF和C-SDF内多糖分子大小不一,受热后碳链和氢键断裂[24]。另外,IDF与SDF因组成、分子量不同,导致两者的热稳定性产生差异。在280 ℃后,热稳定性大小依次为SDF>C-SDF>C-IDF>IDF,与DSC实验结果一致。
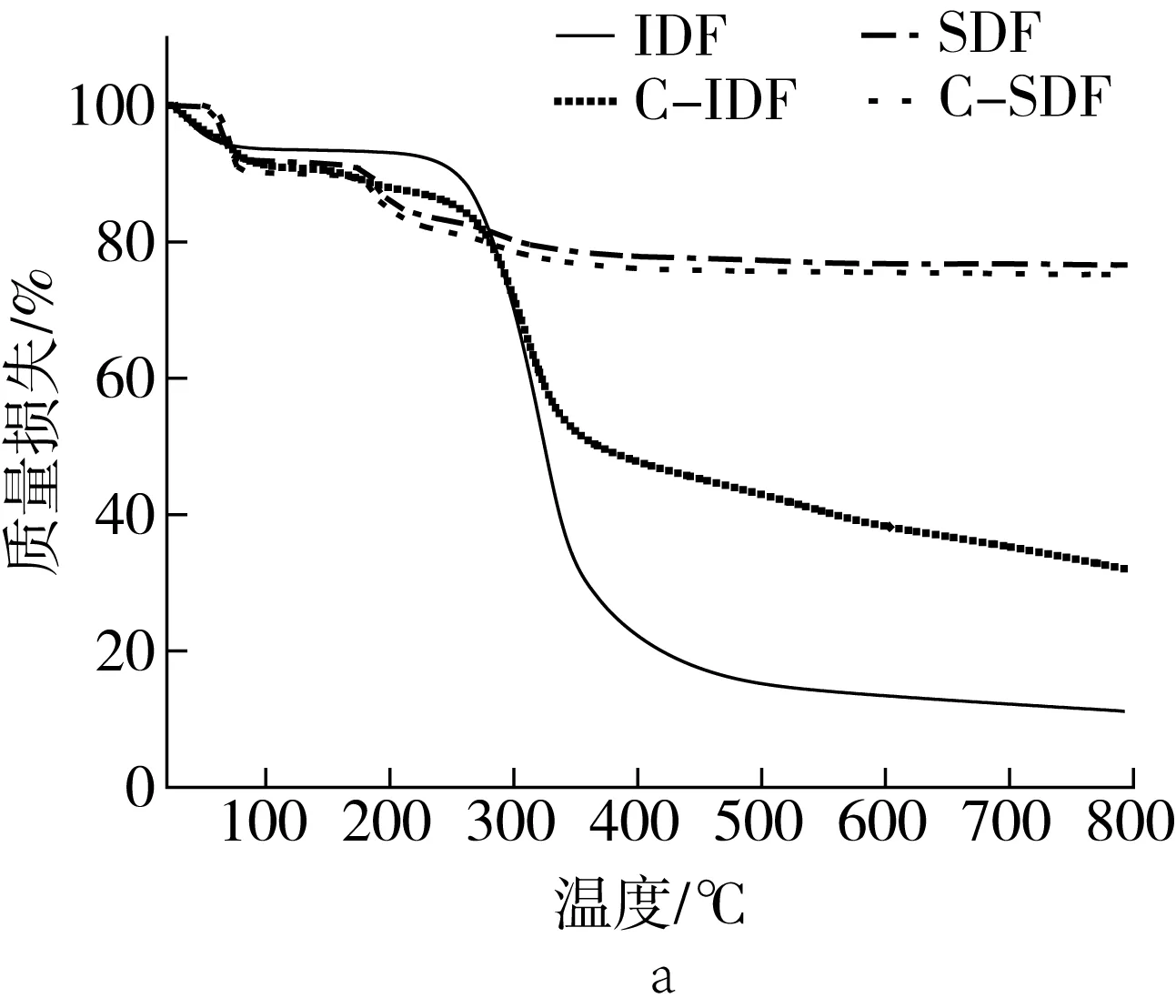
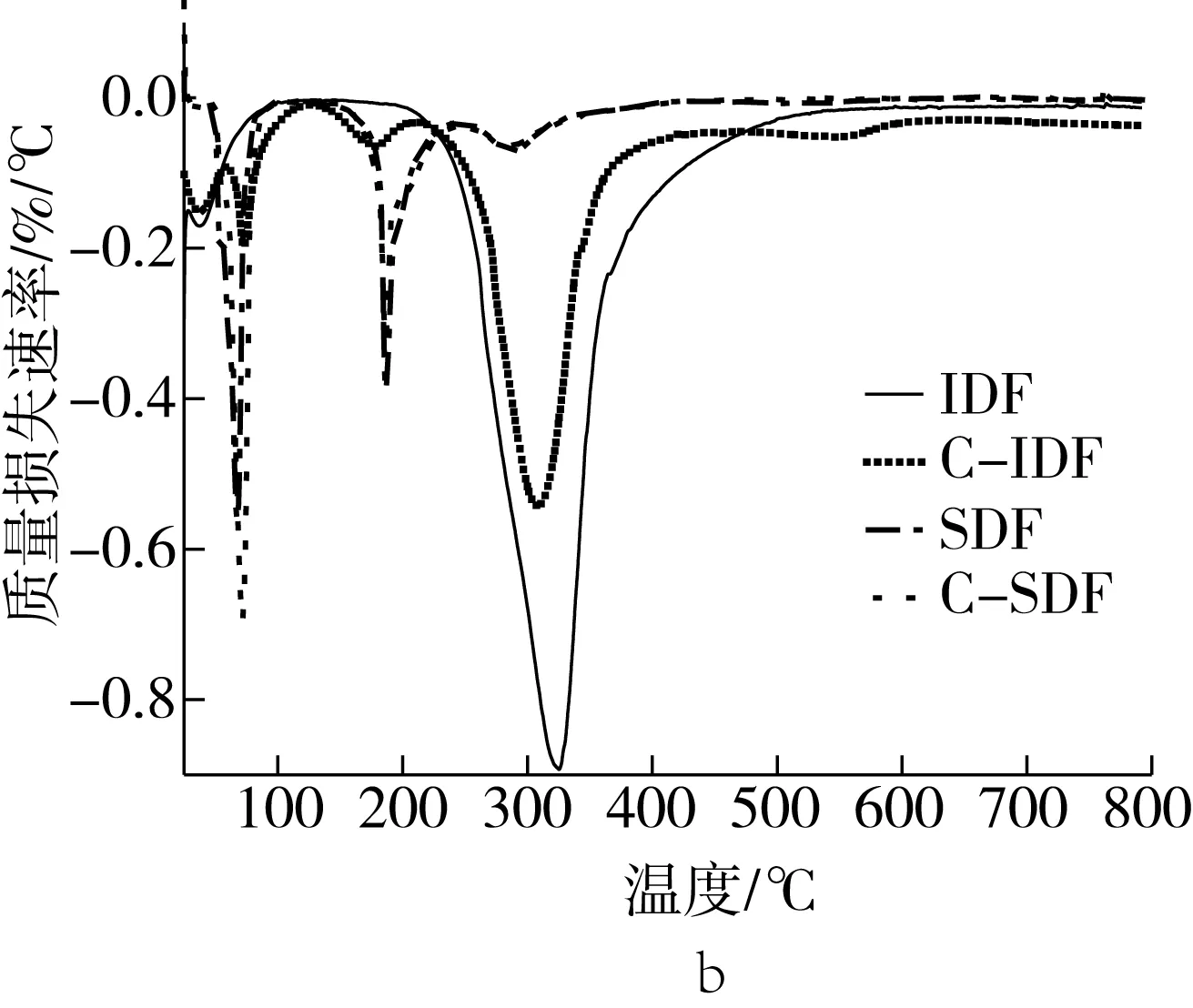
图5 IDF、C-IDF、SDF和C-SDF的质量损失及质量损失速率图
为了进一步阐明IDF和SDF的热转变,进行DSC分析,如图6所示,所有样品在熔化过程均为吸热反应。IDF仅在101 ℃处有1个吸热峰,而C-IDF的第1个吸热峰温度为85 ℃,且向左偏移,第2个吸热峰温度为113 ℃。SDF和C-SDF分别在69、105 ℃和72、100 ℃出现2个吸热峰。说明C-IDF、C-SDF的热稳定性均发生不同程度的改变。图中的吸热涉及水的挥发过程,以及DF和其他多糖的脱甲氧基、脱羟基和脱羧作用[25]。C-IDF和C-SDF的热流强度均低于对照,表明IDF和SDF的热稳定性较低,这与热重结果一致。C-SDF的2个热焓值ΔH(66.51、135.52 J/g)均显著高于对照SDF(39.25、74.22 J/g)(P<0.05),可能是因为游离水和结合水的蒸发,多糖的相变需要更多的热量来破坏氢键以释放水[26]。

图6 IDF、C-IDF、SDF和C-SDF的DSC图
2.3 小米膳食纤维功能性质分析
2.3.1 SC、WHC、OHC分析
由表2可知,与IDF相比,C-IDF的SC增加了9.3%,与SDF相比,C-SDF的SC增加了53.3%;与IDF相比,C-IDF的WHC增加了17.2%,OHC增加了26.9%(P<0.05)。整体上,IDF和C-IDF的SC、WHC、OHC优于SDF和C-SDF,C-IDF的SC、WHC、OHC作用更明显,蒸煮导致部分IDF降解,使IDF的结构发生改变,从而改变IDF的水化性能[27]。IDF和C-IDF的WHC的变化与Wen等[28]研究的酶和酶微化处理对米糠膳食纤维的结果(3.60 g/g)相比明显增加了。DF的OHC对防止食物在烹饪过程中脂肪流失和保持食物风味有重要意义,IDF和SDF可吸收食物中的脂肪来抑制肠道吸收脂肪[29]。蒸煮也能提高IDF和SDF的OHC,该结果与QIAO等[30]的研究结果一致。优质DF具有更高的水化性能,有助于增加饱腹感,从而减少食物摄入量[31]。蒸煮可提高DF的吸附性能,进而提高了对水、油类物质的物理吸附,从而改善DF的SC、WHC、OHC[10]。
2.3.2 胆固醇吸附能力分析
DF可降低小肠对过多的胆固醇、胆酸盐和甘油三酯的吸收和利用,从而降低胆固醇水平[32]。样品与胆固醇(pH 2.0和pH 7.0)的结合能力如表2所示。标准曲线为y=8.377 3x-2.386 5,R2=0.998 8。与IDF相比,C-IDF对胆固醇的吸附量在pH 2.0(模拟胃)环境中增加了10.7%,C-IDF在胃消化阶段对胆固醇吸附量优于肠消化(pH 7.0)阶段,而C-SDF相反,由于在酸性条件下,过多的氢离子阻碍胆固醇携带正电荷[33],导致C-SDF的胆固醇结合能力降低。这是由于蒸煮处理使DF暴露的极性基团增多,空间障碍减少,从而提高胆固醇吸附能力。在胆固醇吸附过程中,DF表面会通过分子间引力形成多分子吸附层,从而对胆固醇起到吸附效果[34]。此外,DF表面的活性基团也可以直接螯合胆固醇分子,进而对胆固醇起到吸附作用[23]。Xu等[35]通过微流化处理桃渣辅助纤维素酶法提取的SDF,显著增强对胆固醇的结合能力。由此可知,DF具有平衡体内胆固醇含量的潜力。
2.3.3 亚硝酸盐吸附能力分析
亚硝酸盐是一种活性离子,可与仲胺和酰胺在酸性条件下反应形成N-亚硝基化合物,在动物体内具有致癌性[36],因此,降低亚硝酸盐含量对于人体具有重要意义。亚硝酸盐吸附标准曲线y=0.839 2x-0.006 5,R2=0.992 9。由表2可知,不同模拟环境下,IDF、C-IDF、SDF、C-SDF对亚硝酸盐的吸附不同,模拟胃中DF对亚硝酸盐的吸附量均高于模拟肠道中的吸附量。这一结果与MOLLER等[37]的结果一致。这是由于蒸煮处理使物料粒径减小,表面积增大,更多的多孔网络结构和官能团暴露[38]。C-IDF吸附亚硝酸盐量较IDF在同一条件下增加了2.8%(pH 2.0)和3.7%(pH 7.0),C-SDF吸附亚硝酸盐量较SDF在同一条件下增加了2.7%(pH 2.0)和10.3%(pH 7.0)(P<0.05),这与蒸煮破坏颗粒完整性有关。DF对亚硝酸盐的高清除能力对预防胃癌发生有重要作用,也作为预防胃癌的功能性食品提供了潜在的应用前景。

表2 IDF、C-IDF、SDF和C-SDF的功能特性参数
3 结论
采用蒸煮对小米进行改性,辅助酶法提取小米膳食纤维,比较了蒸煮前后DF的结构和功能特性。结构特性研究结果表明,蒸煮处理会破坏DF颗粒完整性,使得IDF孔洞变多、SDF出现团聚体;蒸煮处理不影响DF的红外吸收峰,但会使部分水溶性多糖降解,导致晶体结构由有序向无序转变。蒸煮处理还会使DF热稳定性增大、颗粒粒径减小且分布集中。功能特性结果表明蒸煮处理显著提高了IDF的水化特性。模拟消化实验表明蒸煮处理可以改善DF对亚硝酸的吸附能力,这对于预防胃癌发生积极的作用。
猜你喜欢
思维与智慧·下半月(2022年5期)2022-05-17 00:54:54
现代畜牧科技(2021年6期)2021-07-16 05:50:28
纺织科技进展(2021年3期)2021-06-09 08:07:14
陶瓷学报(2021年1期)2021-04-13 01:33:02
当代水产(2019年6期)2019-07-25 07:52:16
读友·少年文学(清雅版)(2018年7期)2018-11-16 03:04:54
当代水产(2018年12期)2018-05-16 02:49:52
小学生优秀作文(低年级)(2017年9期)2017-08-07 02:14:17
创新作文(小学版)(2017年34期)2017-04-09 06:15:20
兽医导刊(2016年12期)2016-05-17 03:51:46
