
手性叔丁基亚磺酰胺的制备方法及其在化学药物合成中的应用价值
2022-01-27 04:31刘文杰谢琪
当代化工研究 2022年2期
*刘文杰 谢琪
(江苏天士力帝益药业有限公司 江苏 223002)
现阶段,随着科学技术的不断发展,手性叔丁基亚磺酰胺在新型医药合成研究中的应用也越来越广泛,是手性胺类药物的重要手性源。在药物合成应用中,手性叔丁基亚磺酰胺的特征在于:(1)能够有效地和醛类等物质发生反应形成亚胺;(2)该辅助药物还具备一定的活化作用,形成的亚胺类产物亲电性较强,后续反应的非对称选择性将得到明显提升;(3)产品的制备过程中,对产物的保护效果较优越,且对于强碱和过渡金属的耐受能力较强;(4)在酸的作用下,非常易离去,经反应获得中间体后再经处理,得到的成品收率较高;(5)合成方法经济;(6)应用的领域较大。
1.亚磺酰胺
在自然界及日常生活中,含氮有机化合物的存在范围非常广泛,且在大多数情况下,都具有一定的生物活性和药用价值,研究发现,大多数化合物的药物活性都和手性胺官能团有关。鉴于此,对于手性胺类化合物而言,其不对称合成的应用则较为重要。现阶段,手性亚磺酰胺属于新型医药中间体,同时也是合成手性胺类药物的关键手性来源,得到了广大研究人员的高度关注。当前,比较熟知的手性亚磺酰胺主要包括叔丁基亚磺酰胺和对甲苯亚磺酰胺,其中叔丁基亚磺酰胺及其衍生产物不但被有效的应用于合成手性药物,同时其也能够当做手性配体诱导催化不对称合成反应。
2.手性的重要性
在自然界中,手性分子的种类非常多,其中大部分的手性分子都有一定的生物活性,由于手性结构的差异性,不同的对映体在生物体中也会呈现出不同的生理活性。手性分子能够有效的和生物大分子进行结合,发生较为复杂的生理作用,并呈现出各类药理及毒理效应。目前,在国内药物销售市场中,大多数的药物均具有手性,其在半合成药物中的占比达到了90%,而在全合成药物中占比也达到了50%,在没有深入研究发展之前,手性药物的不同效用没有得到人们深刻的认识。随着时代的发展,在20世纪60年代,联邦德国药厂研发了镇静催眠药物-沙利度胺。该药物对孕妇的妊娠反应有一定的抑制作用,孕妇服用该药物后,其妊娠反应会得到缓解或消失,故该药物也别称为“反应停”。广泛的用药过程中发现,部分孕妇服用此类药物后,分娩后的婴儿是畸形的,这给家庭和社会带来了巨大的灾难。为了解决这一问题,研究学者发现,R-型的沙利度胺能够控制孕妇呕吐现象,但S-型的沙利度胺可能会导致胎儿畸形,其毒副作用较强。
当沙利度胺事件发生以后,很多国家对具有对映异构体结构的药品,加大了监管、审查力度。在1992年,美国食品药品监督管理局就手性药物的监察标准,颁布了相应的制度要求,随之欧联体、加拿大也相继颁布了相关条例,主要内容是在研发过程中,需对手性药物不同异构体的生理活性、药理作用、毒性进行严格的试验,如果某一对映异构体具有明显的毒副作用,却没有治疗作用,那就需要对其含量做好把控。现阶段,我国对手性药物也进行了严格的规范要求。
3.手性及手性分子简介
在化合物的分子属性中,手性是其中较为重要的一项属性,手性这一名词常出现在化学实验和医药研发领域中。大多数情况下,当一个分子中碳原子上有四个不同结构的基团时,使得该分子无法和其镜像进行有效的重合,我们就将这类分子称之为手性分子,并按一定的规则将其分为(R,S)或(D,L)构型。手性化合物如同人的左右手,尽管是非常的相似,但是在特定的情况下,还是会产生不同的效果,手性分子的应用主要体现在药物、生物、农业等科技领域。通过药物-受体间的结合作用,研究人员可更深刻、形象的了解这一实际状况,因为生物靶点主要以手性实体为主,不同的对映体药物会与其进行选择性结合,而发挥不同的效用。不同对映体存在不同效用的现象比较多见。比如,在20世纪中期,欧洲的“反应停”事件及利尿药茚达立酮,因其结构的立体差异性,所呈现的药效及毒性也是完全不一样的。因此,在药物研发过程中,需要结合药物的结构和药效,对药品内异构体的比例进行科学试验,从而获得更好的治疗效果。
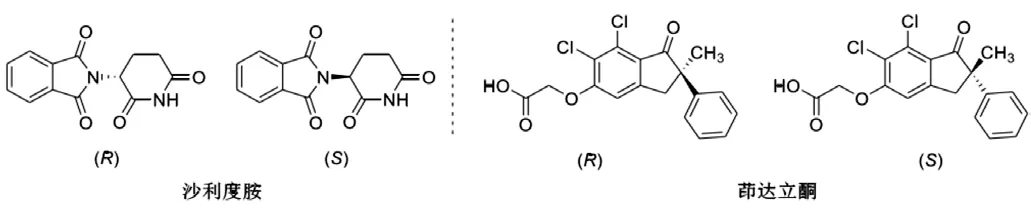
图1 不同构型的药物表现出不同的生理活性
现阶段,随着药物研发水平的不断进步,研究人员与相关部门对新型药物的研发及监管力度也不断加大,各个国家在此过程中均有了一致的理念,相对于手性药物的研发,各国也都规划制定了相应的指导原则。在药物研发过程中,不能忽视药物分子的立体异构对药理、毒理及临床疗效的影响,需以不同的角度去细致化的研究分析。
总的来讲,当药物中有手性分子时,其单一的构型应是后期发展的必然趋势。如何得到不同构型的化合物,经过多年的科学研究,已积累出多种方法,主要包括生物提取、手性不对称合成、对映异构体拆分等,其中手性催化剂、金属与手性配体催化的方式,俨然是最经济、有效的方法。
4.手性叔丁基亚磺酰胺的制备
(1)非对称选择性氧化
在叔丁基亚磺酰胺的制备过程中,通常使用的生产工艺为非对称选择性氧化法。在该方法中,分为两个工序,分别是叔丁基二硫醚的不对称氧化、氨基锂亲核取代。第一步,对叔丁基二硫醚进行不对称催化氧化,不但能够高效率地得到叔丁基亚磺酸盐,且产量较高。第二步,中间体叔丁基亚磺酸盐与氨基锂发生亲核取代反应,完成整个R-叔丁基亚磺酰胺的制备。本制备方法采取叔丁基二硫醚为起始物料,该物质是石油化工生产过程的一个副产物,其特征在于产量大、价格便宜,故R-叔丁基亚磺酰胺的整个生产成本较低。在进行选择性非对称氧化反应时,对于催化剂的选择,主要以Schifi碱-钒的复合物为主。而在实际生产过程中,选择性不对称氧化反应之后还需进行蒸馏操作,除去反应过程中产生的构型转换副产物,提高中间体纯度,之后即可进行后续的氨基锂亲核取代反应制备粗品,对粗品进行精制后,可完成整个叔丁基亚磺酰胺的制备,以上两步骤的总收率可达70%以上。
对于非对称选择性氧化法,科学合理的工艺优化,可一定程度上克服以往的两相体系不利于生产的弊端。除此之外,借助缓慢滴加双氧水的方式,还能够缓解生产过程中料液局部高温的问题,同时也可规避催化剂中毒等不利点,反应的实用性、安全性得到提高。
(2)手性辅助物参与的非对称选择性合成
在研究手性亚砜的合成过程中,技术人员也投入了大量的精力及资金对手性叔丁基亚磺酰胺的工艺进行摸索、优化,最终形成了一套由手性亚砜制备手性叔丁基亚磺酰胺技术。在该项技术领域中,手性叔丁基亚磺酰胺的质量可达到光学纯。在研究过程中,手性辅助物通常以双丙酮-d-葡萄糖为基础底物,利用化合物诱导技术,最终完成手性叔丁基亚磺酰胺的制备,并将手性纯度提高至光学级。在不断的探索研究中,又将茚醇加入至手性辅助物的系列中,通过碱催化,与1,3,5-三甲基苯基磺酰氯产生作用,实现叔丁基亚磺酰胺制备。随着该项研究技术的发展,后续也发现了更多的手性辅助物,比如,可利用去甲麻黄碱为辅助物,经一系列化学反应之后,也可实现R-叔丁基亚磺酰胺合成,且此方法的收率可高达85%。
5.手性叔丁基亚磺酰胺在药物合成中的应用
(1)抗帕金森病药物雷沙吉兰
雷沙吉兰的研究资料显示,其为选择性的二代单胺氧化酶-β抑制剂,可通过降低多巴胺的分解而起作用。当前,对于雷沙吉兰的制备方法研究较多。在传统的制备方法中,通常需经过酒石酸来拆分消旋体产物,这种合成方式不但成本消耗较高,且生产效率较低,无法满足当前发展的生产需求。相关技术人员在传统合成方法的基础上,设计了一条新的合成线路,该工艺以1-茚酮与R-叔丁基亚磺酰胺为起始物料,首先形成中间体亚胺,接着通过NaBH4还原反应和脱叔丁基亚磺酰基,制得关键中间体手性胺源,然后再发生取代、成盐反应,最终制得雷沙吉兰。这种新型的合成工艺,操作方便,生产效率高,产品纯度可高达96%以上。
(2)老年痴呆症治疗药物卡巴拉汀
卡巴拉汀在医学临床中,是治疗老年痴呆症的关键性药物。该药物制备的方法是先进行外消旋产物的合成,接着进行手性拆分,最终得到成品。该药物的制备特点与上述的雷沙吉兰相似,成本消耗较大,生产周期漫长,生产效率偏低,对合成工艺中各化学反应的要求较为苛刻,尤其是其中的不对称合成法,在反应期间还有一定的不稳定性,会严重影响产品的质量,这些问题都影响药物的生产成本及产能扩大。
(3)氨曲南母核
氨曲南属于人工合成的单环类抗菌素,对于该药物的制备,通常以L-苏氨酸为起始物料,经多步反应制得。L-苏氨酸首先通过酯化反应、氨解反应、氨基与羟基保护制备关键中间体,再通过磺化反应、环合反应及脱保护基,最终完成目标化合物的制备。该制备方法的操作较为繁琐,比如,在进行酯化反应、氨解反应期间,所需的时间较为漫长;除此之外,在整个制备过程中,需进行氨基和羟基的保护与脱保护,合成的经济性较低,其中,氨基保护剂主要是(Boc)2O或者氯甲酸苄酯,而(Boc)2O或者氯甲酸苄酯的成本较高,上述各弊端均与现阶段的生产需求不符,不利于生产。为了控制成本,并提高生产效率,上海立科药物化学有限公司就该药物的制备方式,申请了专利—氨曲南中间体3-氨基-2-甲基-4-氧代-1-氮杂环丁基磺酸合成法(专利号为101591282A)。在该专利的合成工艺中,以(R,E)-叔丁基亚磺酰胺基乙基亚胺和邻苯二甲酚亚胺乙酰氯为原料,通过碱催化作用,进行[2+2]反应、脱保护反应和磺化反应,完成氨曲南母核制备。该生产工艺的特征在于合成步骤短,通过较短工序就可获得手性源,生产效率较高。
(4)抗肿瘤药硼替佐米
硼替佐米,结构为二肽硼酸盐类似物,属哺乳动物细胞中26S蛋白酶体糜蛋白酶样活性的可逆抑制剂,常用于多发性骨髓瘤患者的治疗。该药物的合成关键是中间体a-氨基硼酸酯的制备,其合成难度系数较高。研究发现,以2-甲基丙烷硼酸为合成原料,经缩合、加成等多步反应可制得该中间体,但成本费用较高,同时,在生产过程中,需要将温度准确控制在70℃至80℃之间,才可顺利完成反应,而相对于反应本身的特点,该操作难以在工业化中实现,故该工艺不适合产业化发展。在2008年,研究人员Beenen等人员就这一问题,研究并设计了一种新型的合成路线。在该合成路线中,以R-叔丁基亚磺酰胺为原材料,借助频那醇硼烷酸酯的不对称加成,完成硼替佐米的关键中间体a-氨基硼酸酯的制备,其中,目标R构型产物的纯度可高达98%以上,其中S构型杂质的比例相对很小,该新型合成技术的工序较少,且对反应的控制要求不高,以较低的成本可制备出质量更好的成品,适合产业化发展。
(5)左旋西替利嗪
西替利嗪在临床中多用于过敏性疾病的治疗,该药物是一种H1-受体拮抗剂,为二代抗组胺类抗过敏药。现阶段,经过大量的研究试验表明,在抗过敏作用中,与西替利嗪相比,西替利嗪左旋异构体的活性要更优越,同时大量临床试验也表明,左旋体所产生的不良反应更少,利于患者服用。随着不断的深入研究,左旋西替利嗪的应用价值已远大于西替利嗪,逐渐成为第三代抗组胺类抗过敏药物。对于消旋体盐酸西替利嗪的合成工艺,最为常见的方法是,利用1-[(4-氯苯基)苯甲基]哌嗪为起始原料,首先与氯乙醇发生取代反应,中间体再与氯乙酸钠发生缩合反应得到油状粘稠物,在完成一系列复杂的处理包括减压蒸馏等方法之后,还要对产物进行拆分,最终得到西替利嗪的左旋体。该工艺在生产过程中,对于设备的要求较高,处理工艺复杂,产业化生产的难度较大,经过大量的科学摸索及试验研究,现阶段已发明了新型的左旋西替利嗪合成方法,在该合成工艺中,以S-叔丁基亚磺酰胺与苯硼酸作为起始物料,在+1价铑催化剂的作用下,先进行酸化反应制得铵盐中间体,该工序的转化率可达96%,同时光学纯度92%以上,铵盐中间体再进行取代、缩合反应,最终可得到左旋西替利嗪。此工艺操作方便,所需反应步骤较少,但催化剂+1价铑的使用量较大,后续研究表明,以R-叔丁基亚磺酰胺为起始物料,同样使用+1价铑催化剂,也可以制得左西替利嗪,工艺的改进之处在于将催化剂的使用由5%降低至1%-2%,大大降低了生产成本,利于工业化生产。
(6)β-内酰胺型紫杉醇侧链前体的合成
紫杉醇是一种二萜类化合物,其特性在于独特的抗癌活性,主要应用于转移性卵巢癌和乳腺癌的治疗。通过研究发现,在紫杉醇的合成过程中,β-内酰胺型紫杉醇侧链前体是关键中间体,其合成方法较多,研究人员经过不断的科学摸索,提出了一种较为新型的合成方法,该方法的主要过程为:首先对羟基乙酸的烯醇化物进行保护,然后与手性R-叔丁基亚磺酰胺和苯甲醛进行反应,获得亚磺酰亚胺的不对称中间体,最后再经反应得到相应的直链型紫杉醇侧链前体,整个过程收率超过了97%,同时ee值也达到了99%。
6.结语
综上所述,在当前发展形势下,手性药物已成为新型化学药物的一个重要的构成部分,在未来具有非常好的发展前景。手性叔丁基亚磺酰胺作为重要的手性胺源,随着药物合成研究的不断深入,其发挥的作用也越来越大,市场前景也将越来越重要。
猜你喜欢
分子催化(2022年1期)2022-11-02
科学导报(2022年41期)2022-07-13
中国药学药品知识仓库(2022年10期)2022-05-29
太原理工大学学报(2022年2期)2022-03-21
化工环保(2021年3期)2021-06-17
汕头大学学报(自然科学版)(2020年4期)2020-12-14
湖南大学学报·自然科学版(2020年2期)2020-04-17
江苏农业科学(2017年20期)2017-11-30
中成药(2017年5期)2017-06-13
化学教学(2016年10期)2016-11-25
