
碳掺杂锐钛矿TiO2的结构和电子性质
2022-01-14 05:32郎秀峰薛佳卉王唯锦
河北科技师范学院学报 2021年3期
郎秀峰,薛佳卉,王唯锦
(1 河北科技师范学院物理系材料模拟与计算研究室,河北 秦皇岛,066004;2 北京理工大学医工融合学院)
二氧化钛(TiO2)具有光催化活性高、化学性质稳定、无毒无臭以及成本低廉的优势,是最具有发展潜力和研究最多的光催化剂[1, 2]。在Fujishima[2]发现TiO2能够光催化分解水成H2和O2之后,人们做了很多的工作来促进TiO2作为半导体的光化学、光催化和太阳能电池的效率。但是,锐钛矿由于其带隙较大(3.2 eV),只能吸收仅占太阳光能5%左右的紫外光,因此它的光催化效率低。目前,金属或非金属掺杂作为将TiO2的光学吸收范围扩展到可见光区的途径引起了各方广泛的关注[3~6]。为了提高二氧化钛在可见光区的光催化效率,人们进行了非金属掺杂改性的大量实验和理论工作[5],推导了非金属掺杂(如N,C,F或S)在改变TiO2的带隙结构以引发可见光活性方面研究和应用。
关于非金属掺杂TiO2,一个重要的研究方向就是掺杂元素在TiO2的化学状态及其与电子性质的关系。例如,Khan等[6]和Barborini等[7]实验报道了在Ti金属板材的火焰热解、TiC粉末的退火和离子辅助电子束蒸发等条件下制备的TiO2中,C以取代O晶格氧的形式存在;而在使用溶胶-凝胶方法制备的TiO2中,C通常是以取代Ti的形式或者处于间隙位置的方式而存在于晶格中[8,9]。这些不同的碳掺杂结构导致实验报道的吸收光谱中呈现出不同的吸收峰,并且不同实验观测到了不同的可见光响应和光催化性能。许多理论工作也报道了C取代O掺杂的TiO2中,C以C原子,CO或C2的形式存在,C取代Ti掺杂和C占据间隙位置的TiO2中,C以CO2,CO3和CO4-型结构存在[8,10~12]。这些理论工作往往只能解释部分实验现象,而且在解释不同的实验现象时还存在一定的争议。例如,实验和理论上关于C以碳酸根离子的形式掺杂的TiO2能否具有可见光光催化活性这个问题就存在很大的争议[8~10]。这些争议的起源在于人们对C掺杂TiO2的结构与其电子性质的内在联系还没有完全理解。因此,深入研究C掺杂剂的化学状态与能带结构之间的内在关系,对于更好地理解C掺杂剂对可见光的吸附和提高TiO2的光催化性能具有重要意义。
基于这些争论,本次研究的工作运用第一性原理计算系统地研究了C取代O或C填隙的方式掺杂钛矿TiO2的几何结构和电子性质。通过分析这些掺杂结构中C的化学状态,并比较这些结构对应的带隙,来证实C取代O掺杂的TiO2能够具有较好的可见光响应。
1 计算部分
TiO2的结构采用了有48个原子的2×2×1体相超晶胞(16个Ti原子和32个O原子)进行模拟。在此基础上,通过将用1个(或2个)C原子取代1个(或2个)O原子模拟了C取代O掺杂锐钛矿TiO2的结构,和将1个C原子放置在TiO2超晶胞的1个间隙位置来模拟C填隙掺杂TiO2的结构。在密度泛函计算的理论框架下,使用自旋极化的广义梯度近似(Perdew-Burke-Ernzerhof, PBE)交换相关泛函加U(U=4.2)的方法来优化上述结构的晶格参数和原子位置。优化过程中,使用投影缀加平面波(Projector Augmented Wave, PAW)方法来处理原子的核与电子之间的作用[13]。其中Ti原子的核与Ar原子电子排布相同,O和C的结构与He原子电子排布相同。在布里渊区,使用400 eV的平面波截断能和3×3×2的Monkhorst-Pack网格来进行积分。自洽场迭代的收敛阈值设定为1×10-5eV,且所有原子的位置进行几何弛豫,直到每个原子上受到的残余力小于0.2 eV/nm才结束。在优化结构基础上,本次研究采用了PBE+U来计算了态密度(Density of states,DOS)。上述模拟计算均是通过基于密度泛函理论的程序(Vienna ab-initio simulation package,VASP)进行的。
2 结果与分析
笔者首先对纯锐钛矿TiO2超晶胞进行了优化。理论计算的晶胞参数为a=0.765 2 nm和c=0.954 6 nm,这与实验测量的参数值a=0.756 4 nm和c=9.502 0 nm相比,在5%误差内,所以理论优化的结构是合理的。在这个结构中,存在2种C—O化学键(赤道C—O键和轴向C—O键),它们的键长分别为0.195 2 nm和0.199 7 nm。它的带隙为2.40 eV,这个值小于实验报道的锐钛矿TiO2带隙(3.2 eV)。计算带隙偏小主要是由于密度泛函理论本身存在的自相关作用所引起的。由于这个工作主要是比较掺杂和未掺杂TiO2的带隙的变化,所以计算带隙偏小对研究掺杂TiO2带隙的变化规律不会有影响。
2.1 C取代O和C填隙掺杂TiO2的结构
对于C阴离子掺杂TiO2,笔者假定了3种初始结构,并对它们进行了优化。首先,当1个C原子被放置在晶格中1个O原子的位置时,优化后的结构见图1(a),笔者将此结构标记为CO。在这个结构中,C同时与3个Ti原子相连接,形成3个C—Ti键,键长分别为0.202 6 nm和0.219 3 nm,其余的O和Ti原子的位置几乎没有改变。其次,当初始结构中C原子摆放在靠近邻近的O原子时,优化得到了1种新的结构(图1(b)),并将其标记为(CO)O。这个结构中,3个C—Ti键的键长分别为0.208 5 nm,0.215 0 nm 和0.205 3 nm。CO和(CO)O两种结构中的3个Ti—C键相对于纯TiO2中相应的Ti—O键都加长了,这与C的电负性比O的电负性差是有关联的。此外,(CO)O结构中出现了新的C—O键,它的键长(0.144 5 nm)接近于C—O单键的长度(0.143 0 nm)。这个键也使得(CO)O结构能量比CO结构的能量降低了0.47 eV,变得更加稳定了。最近的1篇文献报道了高浓度的C掺杂TiO2时,可能出现了C的二聚体的结构,从而使得掺杂的TiO2具有了更好的可见光吸收能力和光催化性能[10]。因此,笔者使用2个C原子取代了晶胞中的2个临近的O原子构建了一个初始结构,优化得到的结构见图1(c),标记为(C2)O。在此结构中,每个C原子与3个Ti原子成键,键长分别为0.211 0 nm,0.224 2 nm和0.203 4 nm,而且2个C原子形成的C—C键键长为0.134 9 nm。通过进一步比较3种结构的形成能发现,无论在O富裕还是Ti富裕的条件下,(C2)O结构的稳定性都比(CO)O和CO结构的稳定性差。
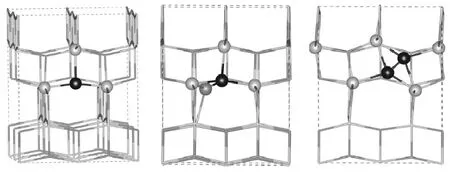
图1 C取代O掺杂TiO2的结构(a)为CO结构,(b)为(CO)O结构,(c)为(C2)O结构;黑色圆球代表C原子,灰色圆球代表Ti原子
笔者将1个C原子置于晶格所有可能的间隙位置进行优化,得到了8种结构(图2)。按照这些结构中所包含的含C原子基团,依次将它们标记为(CO)i, (CO)i-1, (CO)i-2, (CO3)i, (CO3)i-1, (CO3)i-2, (CO3)i-3,和(CO3)i-4。与(CO)O结构一样,在(CO)i, (CO)i-1, (CO)i-2结构中也存在CO的基团(图2(a)~图2(c)),且这些结构中C—O键的长度缩短到0.123 6~0.129 0 nm之间,这接近独立的CO分子中C—O键(0.112 9 nm)。3种结构C—O键的键长差别来源于CO原子基团在结构中的不同取向及C—Ti键数目的不同。这3种结构的能量比较接近,(CO)i-1和(CO)i-2的能量比(CO)i的能量分别高了0.27 eV和0.18 eV。5种含有CO3原子基团的掺杂构型见图3(d)~图3(h)。其中,(CO3)i结构中CO3基团中4个原子不在1个平面上,形成了1个三角锥结构,而在(CO3)i-1,(CO3)i-2,(CO3)i-3和(CO3)i-4中形成平面型CO3基团。进一步观察发现,前面3种结构中平面CO3基团与水平面平行,而最后1种结构中CO3基团与水平面是垂直的。这5种结构中所含的C—O键键长范围为0.128 1~0.144 4 nm,接近于碳酸根离子中3个C—O键的键长(0.136 0 nm)。这5种结构中,(CO3)i-3的能量最低,接近于(CO)i的能量;其它4种结构的能量比(CO3)i-3的能量高了大约0.11~0.25 eV。上述结果充分说明了以CO或者CO3形式进行填隙掺杂的TiO2在能量上是比较接近的,室温下它们都可能实现互相转换。由于这些结构的相似性,笔者在下面的研究中只选取了两种结构讨论它们的电子性质。

图2 C填隙掺杂TiO2的结构(a),(b),(c),(d),(e),(f),(g)和(h)分别为(CO)i,(CO)i-1,(CO)i-2,(CO3)i,(CO3)i-1,(CO3)i-2,(CO3)i-3和(CO3)i-4结构;其中黑色圆球代表C原子,灰色圆球代表O原子
2.2 C取代O和C填隙掺杂TiO2的电子性质
在探讨碳掺杂对TiO2电子性质的影响之前,笔者首先计算了无掺杂TiO2的能带结构、总态密度和局域态密度。结果表明,价带顶部主要由O-2p轨道贡献,导带底部主要具有Ti-3d特征。在此基础上,笔者计算了CO结构的能带结构和态密度。能带结构和态密度都证实了此结构的带隙为2.38 eV,十分接近无掺杂TiO2结构的带隙值(图3)。在带隙中存在3个杂质能级,它们处于费米能级之下,所以它们是被电子占据的。因为这些杂质能级的存在有利于低能量的光将电子激发到导带上,所以它们的存在增加了TiO2对可见光的响应。局域态密度进一步说明了,CO结构中价带顶部和导带底部仍然是由O-2p和Ti-3d轨道来分别贡献的,而杂质能级是C-2p,O-2p和Ti-3d来共同贡献的。
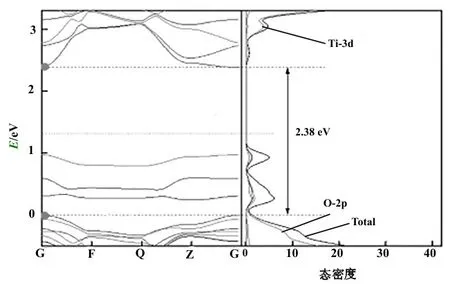
图3 CO能带结构及态密度最上面虚线为导带低所在位置,中间虚线线为费米能级所在位置,最下面虚线为价带顶所在位置
(CO)O和(C2)O结构的态密度见图4(a)和图4(b)。两种结构的态密度图与CO的结构很相似。与未掺杂TiO2结构相比,(CO)O结构的带隙变宽了,而(C2)O的带隙几乎没有变化。两种结构中都存在电子占据的杂质能级,有利于可见光吸收。另外,与CO的局域态密度相似,这些结构的局域态密度图也清晰地说明了这些结构的价带顶部和导带底部仍然是由O-2p和Ti-3d轨道来分别贡献的,而杂质能级是C-2p,O-2p和Ti-3d来共同贡献的。

图4 碳取代氧掺杂TiO2的总态密度注: (a)和(b)分别为(CO)O和(C2)O结构的总态密度和各原子(Ti,O和C)的局域态密度
最后,笔者计算了8种C填隙掺杂的结构中(CO)i和(CO3)i-4的态密度(图5)。其它结构的态密度图与这2种结构的态密度图相似,但是它们的带隙比这2种结构的带隙大很多。(CO)i和(CO3)i的带隙分别为2.46 eV和2.48 eV,略大于无掺杂TiO2的带隙。同时在它们的带隙中也存在电子占据的杂质能级,而且与CO,(CO)O和(C2)O的态密度相比,这些结构的杂志能级更接近导带底部,这更有利于可见光的吸收。局域态密度图进一步说明,这2种结构的价带顶部和导带底部的轨道贡献与其它掺杂结构一样的,但是它们的杂质能级主要来自O-2p轨道和Ti-3d轨道的贡献,没有C-2p轨道的贡献。

图5 碳填隙掺杂TiO2的态密度(a)和(b)分别为典型的(CO)i和(CO3)i-4结构的总态密度以及各原子(Ti,O和C)的局域态密度
以前的研究已经报道了C取代O和C填隙掺杂锐钛矿TiO2的结构,并且调查了它们的电子密度[14]。这个研究只报道1种C取代O掺杂结构和1种C填隙的掺杂结构,它们与上面描述的CO结构和(CO3)i结构很相似。而且这个研究还给出了这2种掺杂构型的能带结构的示意图,它们与CO和(CO3)i结构的态密度是一致的。但是本次研究中描述的其它的掺杂构型及其态密度并没有在这个文献中报道。因此,本次研究有助于人们更全面地认识C掺杂TiO2的结构和电子性质。
3 结论与讨论
本次研究通过第一性原理计算系统研究了以C取代O和C填隙2种方式掺杂的TiO2构型和电子性质。优化得到了3种C取代O和8种C填隙的能带结构。在C取代O的掺杂构型中,C是以C原子,类CO基团和C的二聚体的化学状态存在的,而在C填隙的掺杂构型中,C是以类CO或者类CO3物质的化学状态存在的。进一步计算这些构型的态密度发现,2类掺杂TiO2带隙相比于未掺杂TiO2带隙来说,变化很小,并不能促进TiO2的光吸收范围向可见光区移动。但是这些结构的带隙中都存在电子占据的杂质能级,这对于TiO2吸收可见光是非常有利的。因此,C取代O或C填隙掺杂是可以促进TiO2的光吸收性能的。需要说明的是杂质能级的存在不利于电子或者空穴的迁移,所以它们可能会损坏TiO2的光催化性能。
猜你喜欢
舰船科学技术(2022年21期)2022-12-12
四川大学学报(自然科学版)(2022年4期)2022-07-22
物理学报(2022年11期)2022-06-18
原子与分子物理学报(2022年3期)2022-03-05
山西大学学报(自然科学版)(2021年5期)2021-12-25
物理学报(2019年23期)2019-12-16
青岛大学学报(工程技术版)(2019年2期)2019-09-10
科技创新与应用(2018年21期)2018-09-14
振动工程学报(2017年4期)2018-05-31
电子制作(2018年1期)2018-04-04
