
两个三(邻溴苄基)锡羧酸酯的合成、结构及抗肿瘤活性
2022-01-14 11:30张复兴何丽芳李达伟曾维鸿江叔沄盛良兵朱小明
无机化学学报 2022年1期
刘 熙 张复兴 何丽芳 李达伟 曾维鸿 江叔沄 贺 霞 盛良兵 朱小明
(衡阳师范学院化学与材料科学学院,功能金属有机化合物湖南省重点实验室,金属有机新材料湖南省普通高等学校重点实验室,衡阳 421008)
国家癌症中心最新发布的全国癌症统计数据显示,近10年来,我国恶性肿瘤发病率每年增幅达3.9%,死亡率每年增幅达2.5%,恶性肿瘤占国民全部死因的23.9%[1]。药物治疗已成为当今临床治疗癌症的重要手段之一。近年来,抗肿瘤药物的研发虽然取得了许多重要进展[2⁃3],但仍然缺乏高效、特异性强的抗肿瘤药物。因此,新型抗肿瘤药物的研发对癌症的治疗具有重大意义。有机锡化合物具有良好的抑制癌细胞增殖活性的作用,为开发高选择性、高效、低毒的抗肿瘤药物开辟了新的方向,引起了人们极大的兴趣[4⁃6]。研究表明,许多有机锡化物具有极其高效广谱的抗癌活性[7⁃12],有机锡羧酸酯是有机锡化合物中最重要的一类,不仅具有良好的性能,也具有丰富多变的结构,因而备受人们广泛关注[13⁃19]。为了更系统地研究该类化合物,我们合成了三(邻溴苄基)锡噻吩2⁃甲酸酯(1)和三(邻溴苄基)锡肉桂酸酯(2),通过元素分析、红外光谱、核磁共振谱(1H、13C和119Sn)进行了表征,用X射线单晶衍射测定了晶体结构,对其结构进行量子化学从头计算,探讨了化合物分子的稳定性、分子轨道能量以及一些前沿分子轨道的组成特征。并研究了其热稳定性和体外抗癌活性。
1 实验部分
1.1 仪器与试剂
合成反应在微波有机合成系统(MicroSYNTLab⁃station for Microwave assisted,意大利)中完成。红外光谱用Shimadzu FTIR8700(KBr压片,400~4 000 cm-1)光谱仪测定。元素组成用PE⁃2400型元素分析仪测定,核磁共振用Avance Ⅲ HD 500 MHz全数字化超导核磁共振谱仪(瑞士Bruker公司,TMS为内标)测定。高分辨质谱(HRMS)用Waters Acquity UPLC H⁃CLASS XEVO G2⁃XS Qtof飞行时间串联液质联用仪测定。1和2的晶体结构用BrukerSmart Apex Ⅱ CCD单晶衍射仪测定。1和2的熔点用北京泰克X⁃4数字显微熔点仪测定。热稳定性分析利用TG209F3热分析仪进行,在空气气氛中,加热速度为20 ℃·min-1,气体流速为20 mL·min-1,在40~800 ℃范围内对1和2进行热重测试。所有试剂为分析纯。
1.2 化合物的合成
化合物 1:取 0.708 g(1 mmol)三(邻溴苄基)溴化锡、0.128 g(1 mmol)噻吩 2⁃甲酸和 1 mmol三乙胺溶于40 mL甲醇中,将溶液转入聚四氟乙烯微波反应罐中,将罐盖密封好,置于微波反应器中。设置微波有机合成系统温度120℃,微波辐射反应2 h。冷却过滤,除去不溶性固体,滤液旋转蒸发除去部分溶剂,放置析出白色固体,用二氯甲烷-甲醇重结晶得无色透明晶体0.516 g,产率68.26%。熔点:89~91℃。元素分析按C26H21Br3O2SSn的计算值(%):C,41.27;H,2.78。实测值(%):C,41.13;H,2.72。IR(KBr,cm-1):3 082,3 053,2 979,2 932ν(C—H),1 614νas(COO-),1 361νs(COO-);538ν(Sn—C),426ν(Sn—O)。1H NMR(CDCl3,500 MHz):δ7.69(dd,J=3.5 Hz,J=1.0 Hz,1H),7.53~7.46(m,4H),7.16~7.13(m,3H),7.10~7.07(m,4H),6.96~6.93(m,3H),2.93(s,6H)。13C NMR(CDCl3,125 MHz):δ191.85,139.21,135.29,133.83,132.08,130.13,129.81,128.77,127.56,126.44,123.58,28.92。119Sn NMR(CDCl3,186 MHz):δ5.61。HRMS(ESI)m/zC26H21Br3O2SSn[M+Na]+计算值:778.770 6,实测值:778.770 8。
化合物2:按照上述方法,用0.148 g(1 mmol)肉桂酸代替噻吩2⁃甲酸,得无色透明晶体三(邻溴苄基)锡肉桂酸酯0.594 g,产率76.54%。熔点:107~108℃。元素分析按C30H25Br3O2Sn的计算值(%):C,46.39;H,3.22。实测值(%):C,46.67;H,3.28。IR(KBr,cm-1):3 057,3 042,2 982,2 929ν(C—H),1 643νas(COO-),1 337νs(COO-),554ν(Sn—C),434ν(Sn—O)。1H NMR(CDCl3,500 MHz):δ7.60(d,J=16 Hz,1H),7.56~7.54(m,2H),7.50~7.48(m,3H),7.43~7.39(m,3H),7.19~7.1(m,3H),7.12~7.08(s,3H),6.99~6.93(s,3H),6.52(d,J=16 Hz,1H),2.91(s,6H)。13C NMR(CDCl3,125 MHz):δ191.74,144.68,139.43,134.78,132.07,130.11,129.88,128.75,127.94,127.51,126.33,123.56,119.17,28.78。119Sn NMR(CDCl3,186 MHz):δ-3.30。HRMS(ESI)m/zC30H25Br3O2Sn[M+Na]+计算值:798.829 8,实测值:798.827 5。
1.3 晶体结构测定
分别选取大小为0.21 mm×0.20 mm×0.17 mm(1)和0.23 mm×0.21 mm×0.20 mm(2)的晶体,在Bruker SMART APEX Ⅱ CCD单晶衍射仪上,采用经石墨单色化的MoKα射线(λ=0.071 073 nm),在296(2)K下以φ⁃ω扫描方式收集数据。可观察衍射点数分别为4 672和6 621[I>2σ(I)],用于结构分析和精修。衍射强度数据经多重扫描吸收校正,晶体结构中大部分非氢原子由直接法解出,其余部分非氢原子在随后的差值傅里叶合成中陆续确定,对所有非氢原子坐标及其温度因子采用全矩阵最小二乘法精修。由理论加氢法给出氢原子在晶胞中的位置坐标,对氢原子和非氢原子分别采用各向同性和各向异性热参数精修,全部结构分析工作在WINGX上调用SHELX⁃97程序完成。化合物1和2的主要晶体学数据列于表1。
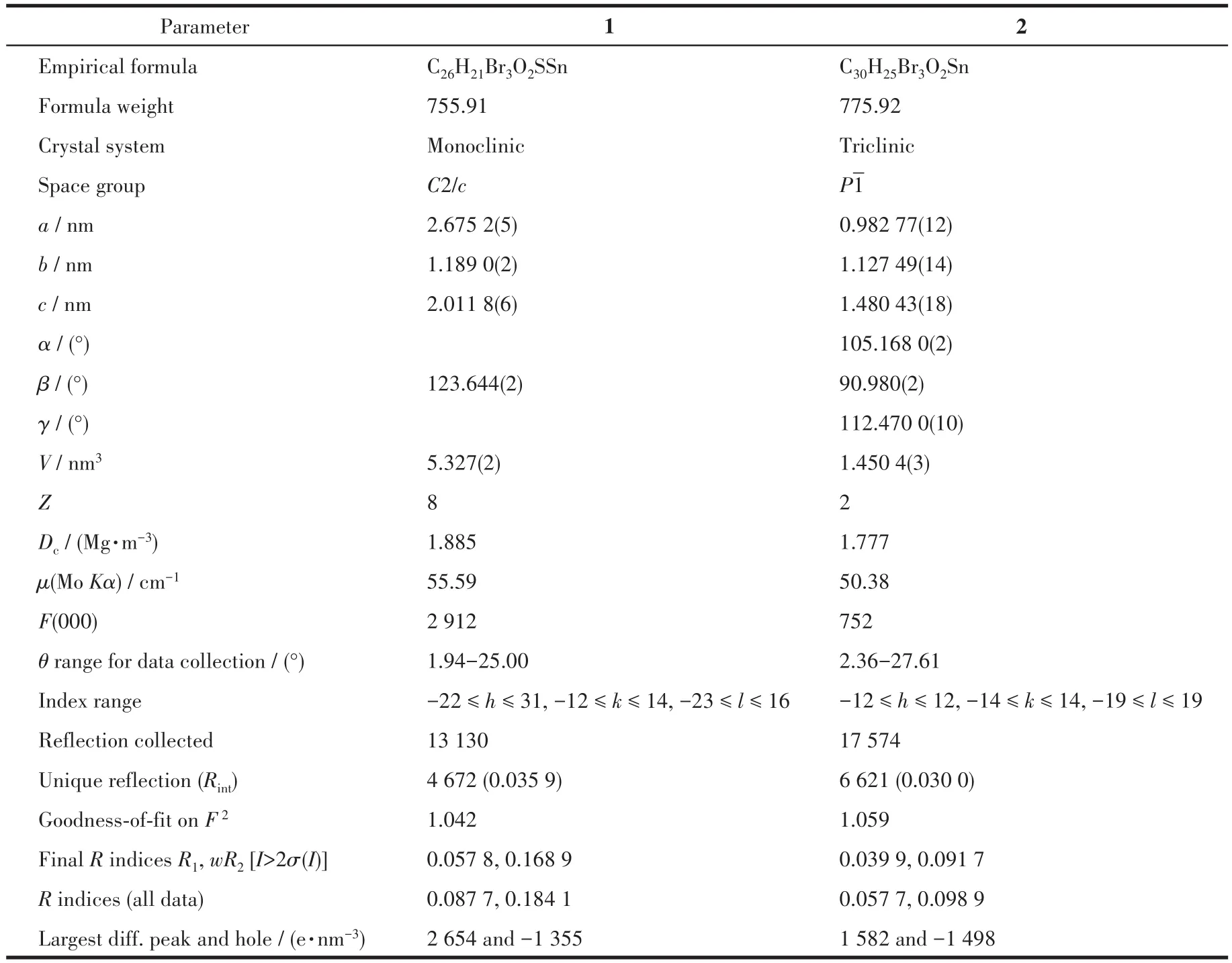
表1 化合物1和2的晶体学数据Table 1 Crystallographic data of compounds 1 and 2
CCDC:2109938,1;2109939,2。
1.4 化合物的体外抗癌活性测定
人乳腺癌细胞(MCF⁃7)、人非小细胞肺癌细胞(A549)和人大细胞肺癌细胞(H460)取自美国组织培养库,用含10%牛胎血清的RPMI1640(GIBICO,Invitrogen)培养液,在含体积分数5%的CO2的培养箱内于37℃下培养,用MTT法检测细胞增殖与生长抑制情况,调整实验细胞数量使在570 nm获得1.3~2.2的吸光度,将化合物1和2的测试药液(0.1 nmol·L-1~10 μmol·L-1)设置6个浓度,处理细胞72 h,每个浓度至少进行3个平行和3次重复实验,应用Graph⁃Pad Prism5.0软件统计分析确定IC50值。
2 结果与讨论
2.1 化合物的谱学性质
在IR中,化合物1在1 614和1 362 cm-1处出现的尖锐吸收峰为羧基的反对称伸缩振动(νas,COO)和对称伸缩振动吸收峰(νs,COO),而化合物2的νas,COO和νs,COO分别在1 643和1 337 cm-1,其 Δν(Δν=νas,COO-νs,COO)分别为252和306 cm-1,均大于200 cm-1,表明化合物中羧基氧都是以单齿形式与锡配位。化合物1和化合物2分别在427和434 cm-1处出现了吸收峰,该峰为Sn—O键的伸缩振动吸收峰,说明化合物中有Sn—O键存在。
在1H NMR谱中,化合物1和2分别在化学位移7.69~6.93和7.56~6.93之间出现多重峰,对应为芳环上的质子,与锡相连的亚甲基氢的δ分别在2.93和2.91;化合物2的烯烃碳上的2个质子的δ分别在7.60和6.52处出现了双重峰,其偶合常数均为16 Hz。在13C NMR谱中,化合物1和2羰基碳的δ分别在191.85和191.74处,亚甲基碳的δ分别在28.92和28.78处;化合物1芳环上碳的δ出现在139.21~123.85,化合物2芳环上碳和烯烃上碳的δ在144.68~119.17之间。在119Sn NMR谱中,化合物1和2分别在δ为5.61和-3.30处出现峰。以上红外和核磁数据结果与X射线单晶衍射测定的晶体结构相吻合。
2.2 晶体结构分析
化合物的主要键长和键角列于表2,化合物1和2的分子结构见图1、2,晶胞堆积图见图3、4。由分子结构图和结构参数可知:2个化合物均为单锡核结构,中心Sn原子与3个亚甲基C原子和1个羧基O原子相连形成四面体构型。配体和溴代苄基的空
间互斥作用使化合物中3个Sn—C键的键长、键角不等。化合物1和2中Sn原子与2个羧基O原子之间的距离分别为0.207 2、0.277 0 nm和0.206 0、0.277 8 nm,都只有其中的一个小于Sn原子与O原子的共价半径之和(0.216 nm),而另一个远大于这2个原子的共价半径之和,说明2个化合物中都只有一个羧基O原子与Sn很好地成键,而另一个羧基O原子未能与Sn原子成键。因此,2个化合物中的羧基均是以单齿形式与锡原子配位,生成四配位的畸形四面体,与红外光谱测得的结果一致。化合物2与已报道的三(邻氯苄基)锡肉桂酸酯[20]具有相似的晶体结构,两者都属三斜晶系,空间群均为P1,但由于溴原子和氯原子的电子效应和空间效应的差别,使得中心锡原子与配位原子的键长与键角存在差异。
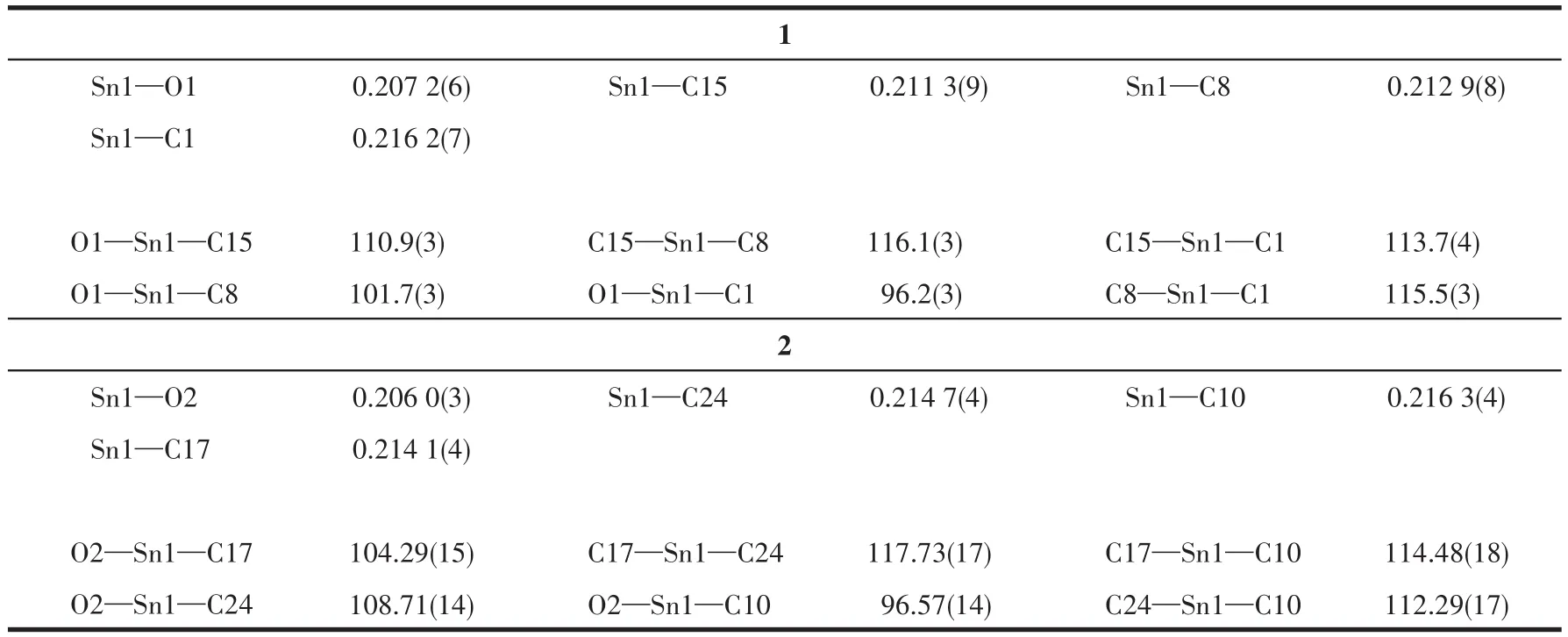
表2 化合物1和2的主要键长和键角Table 2 Selected bond lengths(nm)and bond angles(°)of compounds 1 and 2
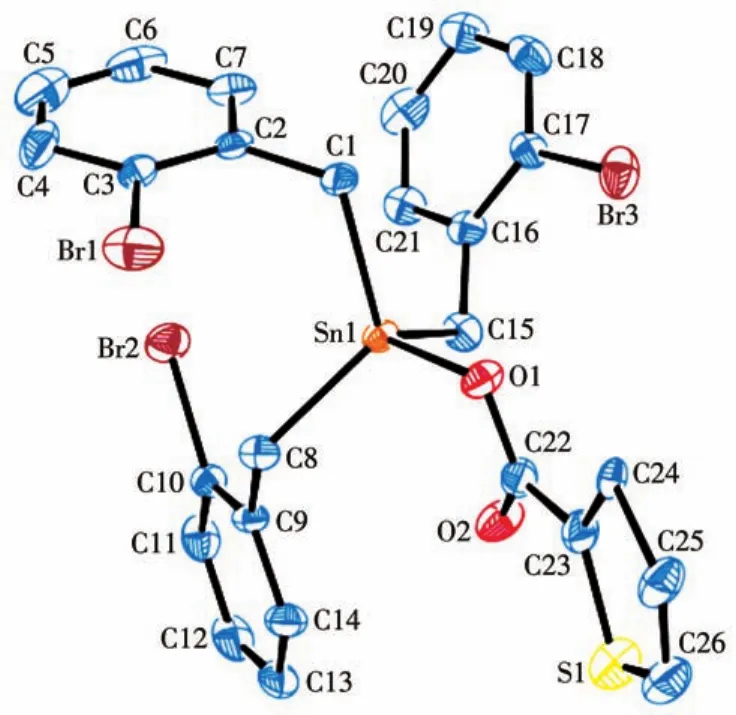
图1 化合物1的椭球概率15%的分子结构图Fig.1 Molecular structure of compound 1 with ellipsoids drawn at 15% probability level
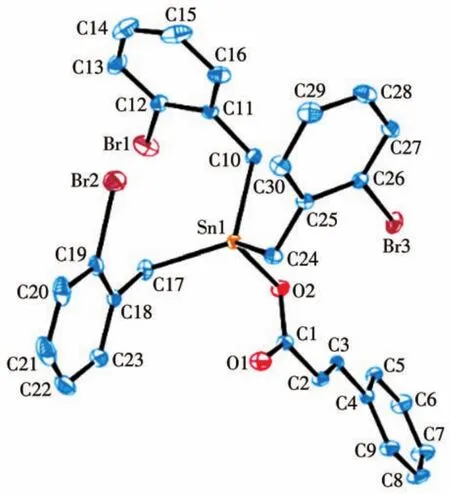
图2 化合物2的椭球概率15%的分子结构图Fig.2 Molecular structure of compound 2 with ellipsoids drawn at 15% probability level
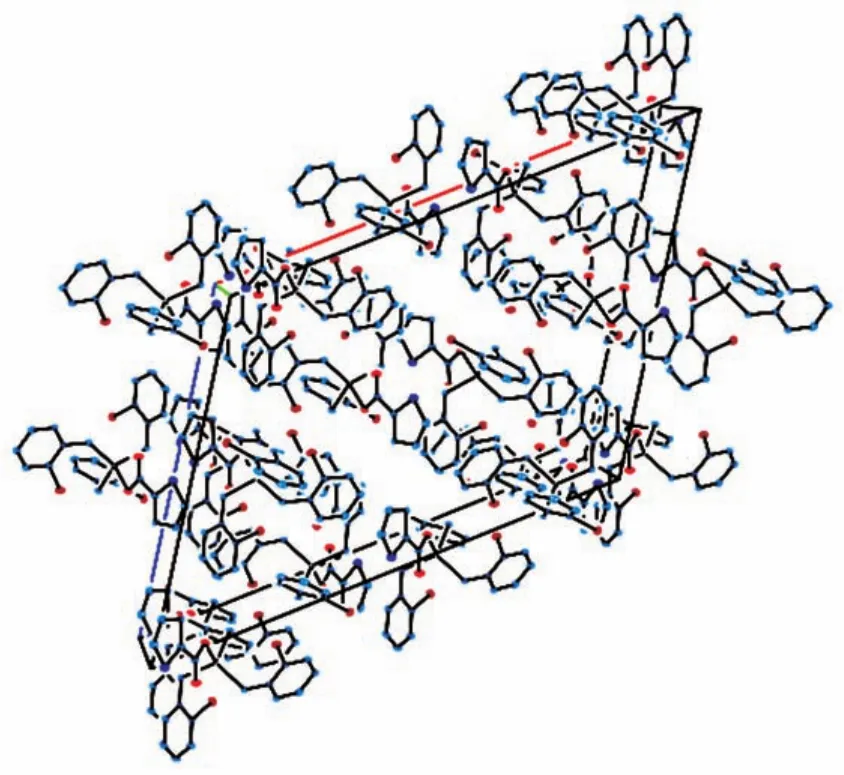
图3 化合物1的晶胞堆积图Fig.3 Packing of compound 1 in a cell
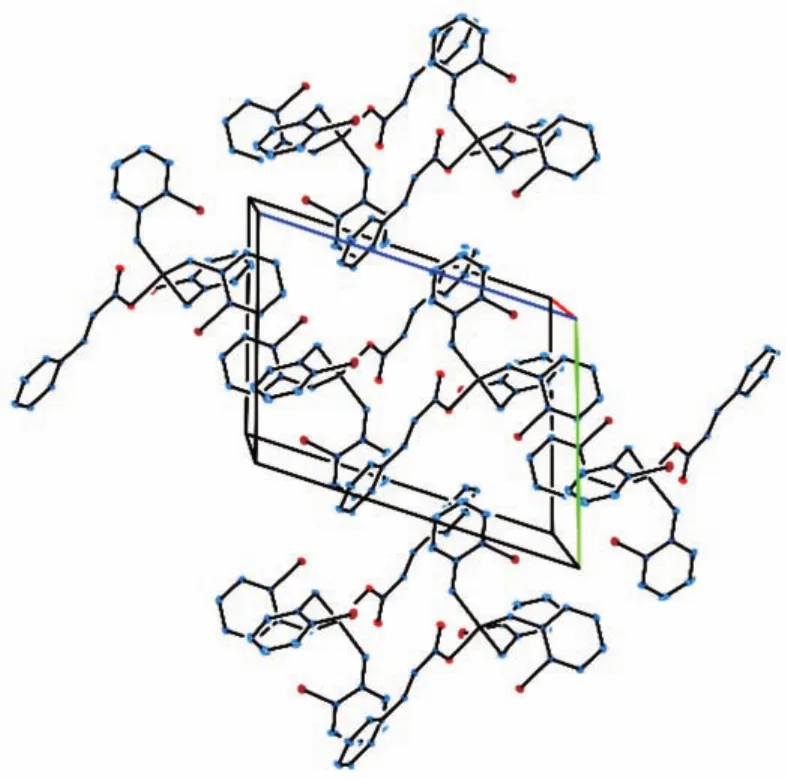
图4 化合物2的晶胞堆积图Fig.4 Packing of compound 2 in a cell
2.3 量子化学研究
根据晶体结构的原子坐标,运用Gaussian 03W程序在B3LYP/lanl2dz基组水平上计算得到1和2的分子总能量和分子轨道能量。化合物1:ET=-1 197.684 165 4 a.u.,EHOMO=-0.311 07 a.u.,ELUMO=0.066 30 a.u.,ΔE=ELUMO-EHOMO=0.377 37 a.u.。化合物2:ET=-1 341.572 632 4 a.u.,EHOMO=-0.314 44 a.u,ELUMO=0.066 94 a.u.,ΔE=0.381 38 a.u.。从体系能量和前沿轨道的能量来看,体系能量和前沿占有轨道的能量均较低,表明1和2分子结构较稳定。最高占据轨道与最低未占轨道的能量间隙ΔE均较大,说明1和2均较难失去电子而被氧化。
为探索化合物1和2的电子结构与成键特征,将化合物的分子轨道进行分析,用参与组合的各类原子轨道系数的平方和来表示该部分在分子轨道中的贡献,并经归一化。分别把化合物的原子分为7部分,化合物1:(a)锡原子Sn;(b)配体羧基碳原子和氧原子L;(c)苄基亚甲基碳C1;(d)配体噻吩环碳和硫M;(e)苄基苯环碳原子C2;(f)溴原子Br;(g)氢原子H。化合物2:(a)锡原子Sn;(b)配体羧基碳原子和氧原子L;(c)苄基亚甲基碳C1;(d)配体苯乙烯基碳原子C2;(e)苄基苯环碳原子C3;(f)溴原子Br;(g)氢原子H。前沿占有轨道和未占有轨道各取5个,计算结果如表3、4和图5、6所示。
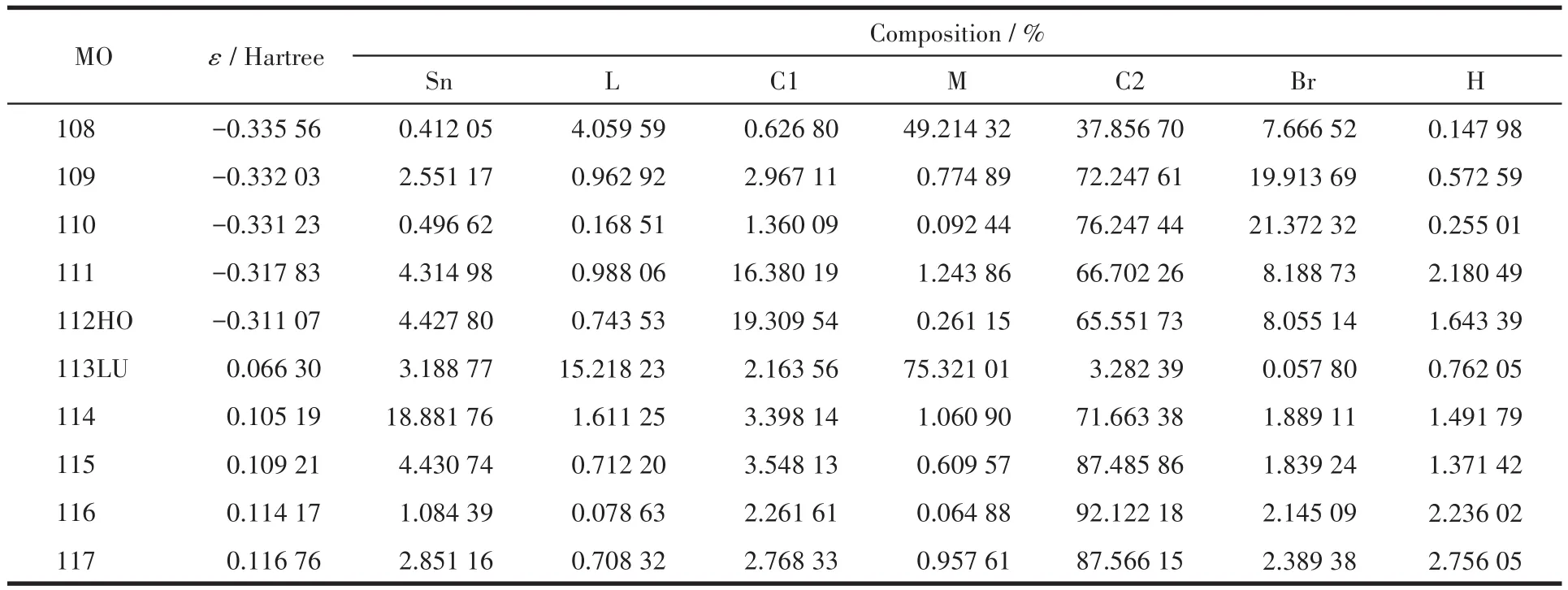
表3 化合物1的前沿分子轨道组成Table 3 Calculated frontier molecular orbital composition of compound 1
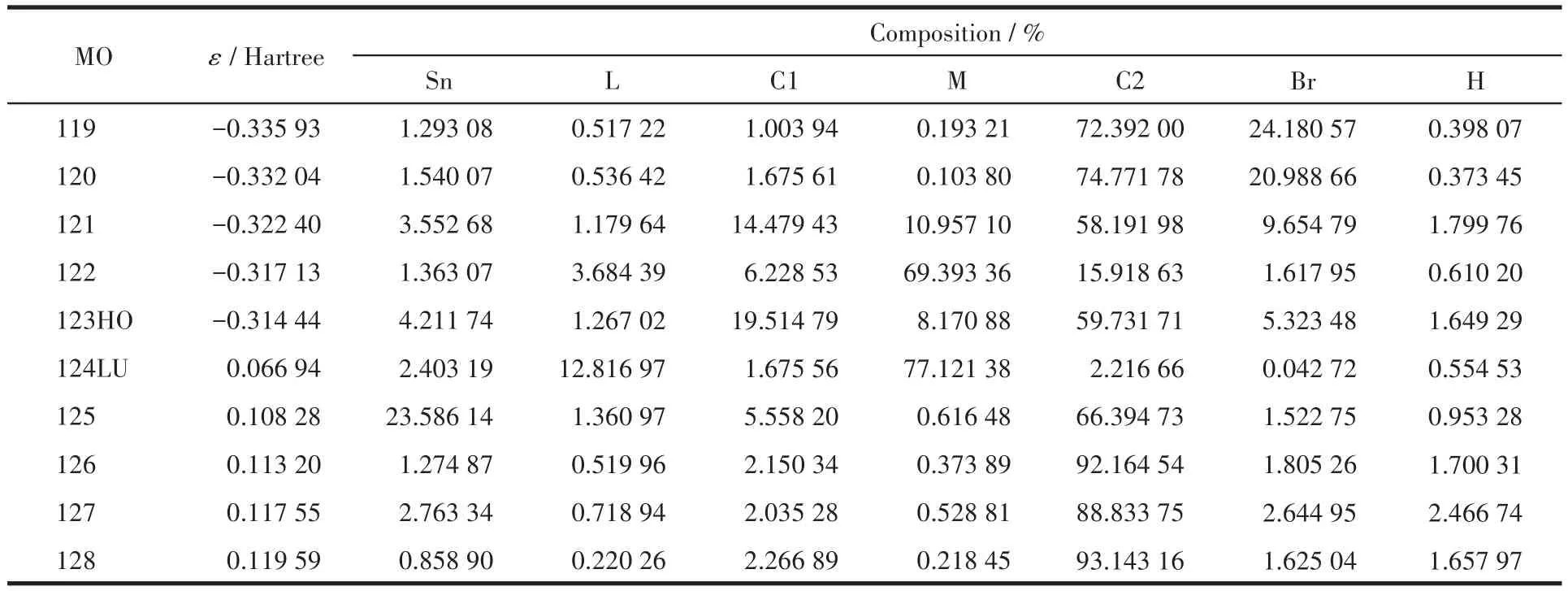
表4 化合物2的分子轨道组成Table 4 Calculated frontier molecular orbital composition of compound 2

图5 化合物1的前沿分子轨道示意图Fig.5 Schematic diagram of frontier molecular orbital for compound 1
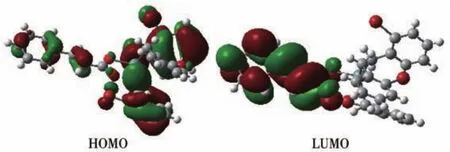
图6 化合物2的前沿分子轨道示意图Fig.6 Schematic diagram of frontier molecular orbital for compound 2
表3和图5显示化合物1的成键特征:前沿占有分子轨道中,对分子轨道贡献最大的是邻溴苄基苯环碳原子和亚甲基碳原子,分别为65.55%和16.38%;其次是溴原子和锡原子,分别为8.06%和4.31%。说明:一是溴代苯环具有良好的共轭性和较强的稳定性;二是亚甲基碳原子与锡原子结合良好,分子中Sn—C键较稳定。比较HOMO与LUMO的各类原子轨道成份,可以看出,当电子从HOMO激发到LUMO轨道时,其它原子上的电子都集中向配体转移,配体的羧基既是电子转移的桥梁也是电子转移的部分受体,而配体的噻吩环则是电子转移的主要受体。
表4和图6显示化合物2的成键特征:前沿占有分子轨道中,对分子轨道贡献最大的是邻溴苄基苯环碳原子和亚甲基碳原子,分别为59.72%和19.51%;其次是配体苯乙烯基碳原子、溴原子和锡原子,分别为8.17%、5.32%和4.21%。说明:一是溴代苯环具有良好的共轭性和较强的稳定性;二是亚甲基碳原子与锡原子结合良好,分子中Sn—C键较稳定;三是配体的苯乙烯基的共轭离域性和稳定性良好。比较HOMO与LUMO的各类原子轨道成份,可以看出,当电子从HOMO激发到LUMO轨道时,其它原子上的电子都集中向配体转移,配体的羧基既是电子转移的桥梁也是电子转移的部分受体,而苯乙烯基则是电子转移的主要受体。
2.4 热稳定性分析
热稳定性分析结果如图7所示。在160℃之前,化合物1几乎没有失重;从160℃起化合物开始缓慢失重,到200℃时化合物开始快速失重,到390℃时失重变缓,至650℃时失重基本停止,残留质量最后稳定在18.28%。总计失重81.72%,残余物可被假定为SnO2,与19.94%的计算值基本吻合。对于化合物2,在170℃之前,其几乎没有失重;从170℃开始化合物快速失重,到380℃时失重变缓,至620℃时失重基本停止,残留质量最后稳定在18.06%。总计失重81.94%,残余物可被假定为SnO2,与19.42%的计算值基本吻合。

图7 化合物1和2的热分析曲线Fig.7 Thermogravimetric analysis curves of compounds 1 and 2
2.5 抗肿瘤活性
测试了化合物1和2对MCF⁃7、A549和H460的体外生长抑制活性,结果见表5。发现化合物1和2对所研究癌细胞均显示了较强的抑制活性。化合物更多的生物活性有待进一步研究
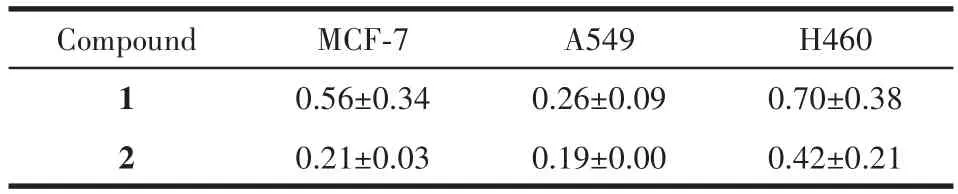
表5 化合物1和2对体外肿瘤细胞的IC50Table 5 IC50of compounds 1 and 2 on tumor cells in vitro μmol·L-1
3 结 论
以甲醇为溶剂,在微波溶剂热条件下,合成了2个三(邻溴苄基)锡羧酸酯:三(邻溴苄基)锡噻吩2⁃甲酸酯和三(邻溴苄基)锡肉桂酸酯。体外抗癌活性测试表明2个化合物对人乳腺癌细胞(MCF⁃7)、人非小细胞肺癌细胞(A549)和人大细胞肺癌细胞(H460)均显示出较强的抑制活性。
猜你喜欢
天津农学院学报(2022年2期)2022-08-05
渤海大学学报(自然科学版)(2022年1期)2022-07-25
云南大学学报(自然科学版)(2022年3期)2022-05-25
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中小学班主任(2019年12期)2019-09-10
新课程·中旬(2016年12期)2017-05-08
中学生数理化·高二版(2017年2期)2017-04-19
科技创新导报(2016年30期)2017-03-15
