
还原氧化石墨烯负载球形金属酞菁电催化阴极氧还原反应性能
2022-01-14 11:29谭阿敏刘布雷杨正超
无机化学学报 2022年1期
谭阿敏 何 洋 刘布雷 杨正超 田 敏 张 改*,
(1西安工业大学材料与化工学院,西安 710021)
(2中国石油独山子石化公司,克拉玛依 833699)
0 引 言
新型清洁能源材料的开发和利用能够有效解决能源枯竭和环境污染问题,对社会经济发展也具有重大的实际意义[1]。直接甲醇燃料电池(DMFC)具有结构简单、能量转化率高、绿色环保等优点而成为新能源研究与开发的热点[2⁃3]。但是,氧电极过电势过高造成的反应速率缓慢等问题,使电池实际能量密度远低于理论值[4]。另外,Pt/C催化剂能有效降低电极过电势,但金属铂价格昂贵,严重制约了DMFC商业化应用[5]。因此,开发新型低成本、高活性的非贵金属催化剂成为提升DMFC综合性能的重要途径。
近年来,人们发现MN4型类卟啉大环化合物具有平面共轭结构和高的还原氧分子活性,在非贵金属催化剂领域受到了广泛关注[6]。酞菁是一种具有类卟啉型18π电子共轭结构的大环分子,具有优异的类血红素载氧功能,能够实现仿生电催化还原分子氧[7]。与卟啉分子相比,酞菁单体由4个异吲哚单元通过亚胺键连接而成,4个苯环周围的氢原子可以由其他原子或基团取代,这种特性赋予酞菁独特的结构可设计性及广阔的应用前景[8⁃9]。研究发现金属酞菁的中心金属原子和MN4型共轭结构是影响其催化活性的主要因素[10⁃11]。其中一种新型双核球形金属酞菁在氧还原反应(ORR)中的出色表现受到广泛关注[12⁃16],其面对面结构单元之间的距离对催化活性影响显著,因此设计、合成不同结构的双核球形金属酞菁对催化ORR具有重要意义。然而,酞菁的18π电子共轭结构使其很容易发生团聚[17],导致催化活性点减少,催化性能显著降低,成为限制其应用的主要不利因素。研究发现,氧化石墨烯(GO)具有较高的比表面积和良好的导电性,是良好的ORR和析氧反应(OER)双功能催化剂之一[18]。在氧气三相催化还原反应中,GO不仅能够高效分散催化剂粒子、降低电极材料电阻,而且还提供三相界面结构,有望解决金属酞菁团聚的问题,提升催化剂的综合性能[19⁃22]。
我们基于金属酞菁及GO的性能特点,通过“π⁃π堆积”作用设计、合成了具有仿生电催化活性的金属酞菁负载还原氧化石墨烯(rGO)复合催化剂M2Pc2(TA)4/rGO(M=Zn、Co、Fe)。利用循环伏安(CV)法和线性扫描伏安(LSV)法探讨了M2Pc2(TA)4/rGO的ORR催化性能及抗甲醇性能,为燃料电池阴极ORR催化剂的研发提供新思路。
1 实验部分
1.1 主要试剂及仪器
试剂包括3,4⁃二羟基苯甲醛(阿拉丁试剂有限公司)、4⁃硝基邻苯二甲腈(阿拉丁试剂有限公司)、N,N⁃二甲基甲酰胺(DMF,南京化学试剂股份有限公司)、Zn(OAc)2·2H2O(上 海 精 细 化 工 有 限 公 司)、Co(OAc)2·2H2O(上海精细化工有限公司)、Fe(OAc)2·2H2O(上海精细化工有限公司)、聚苯乙烯磺酸钠(济南鑫森源化工有限公司)、1,8⁃二氮杂二环[5.4.0]十一碳⁃7⁃烯(DBU,阿拉丁试剂)、二羟基苯甲酸-二(3,4⁃二氰基苯基)醚(自制)、单层 GO(苏州碳丰石墨烯科技有限公司)。
仪器包括X射线衍射仪(XRD,德国布鲁克D2 PHASER型,CuKα辐射源,λ=0.154 2 nm,管电压为40 kV,管电流为100 mA,扫描速度为4(°)·min-1,测试范围 2θ=5°~80°)、红外光谱仪(TENSOR Ⅱ,德国Bruker Optics,测试范围为 3 800~400 cm-1)、紫外可见分光光度计(UV⁃2550,日本岛津公司,溶剂为DMF,测试范围为200~800 nm)、扫描电镜(SEM,Quanta 400F,菲利普电子公司,管压5 kV)、透射电镜(TEM,JEM 2010型,日本电子公司,加速电压100 kV)、X射线光电子能谱仪(XPS,ESCLAB 250,日本电子公司,AlKα光源,电压1 486.6 eV)、电化学工作站(CHI660D,上海辰华仪器有限公司);旋转圆盘电极(AF⁃SMRCE,Pine Instrument Company)、玻碳电极(CHI104,上海辰华仪器有限公司)。
1.2 M2Pc2(TA)4/rGO催化剂的合成
1.2.1 球形金属酞菁M2Pc2(TA)4的合成
二羟基苯甲酸-二(3,4⁃二氰基苯基)醚的制备:将14.48 mmol 4⁃硝基邻苯二甲腈和7.24 mmol 3,4⁃二羟基苯甲醛加入30 mL无水DMF中,于70℃、N2氛围下搅拌2 h。随后,加入7.24 mmol干燥的K2CO3,继续反应120 h。待反应结束,将反应混合物加入到100 mL冰水中,快速搅拌,离心收集沉淀并干燥,得到褐色固体。产量2.600 0 g,产率74%,熔点大于150℃。元素分析按C23H10N4O3计算的理论值(%):C 70.77,H 2.58,N 14.35。实验值(%):C 70.43,H 2.47,N 14.26。IR(KBr,cm-1):2 231(C≡N),1 595(C=C),2 937(Ar⁃CH)。1H NMR(600 MHz,DMSO):δ9.58(s,1H,aldehyde⁃H),8.09~7.98(d,J=7.64 Hz,2H,Ar⁃H),7.95~7.88(d,J=7.52 Hz,2H,Ar⁃H),7.61~7.57(s,1H,Ar⁃H),7.53~7.39(m,4H,Ar⁃H)。
Zn2Pc2(TA)4的制备:将0.77 mmol二羟基苯甲酸-二(3,4⁃二氰基苯基)醚、1.4 mmol Zn(OAc)2·2H2O、0.2 mL DBU、7 mL正戊醇依次加入高压反应釜中,在氮气氛围、180℃条件下反应18 h。冷却至室温后,将混合物加入甲醇中,离心收集沉淀,用甲醇、乙醇、乙醚依次洗涤数次,得到墨绿色粗产物。采用柱层析(V二氯甲烷/V甲醇=8∶2)分离并收集目标产物,旋转蒸发得到0.210 0 g深绿色固体,产率42%,熔点大于300℃。元素分析按C46H24N8O6Zn计算的理论值(%):C 64.99,H 2.84,N 13.18。实验值(%):C 64.87,H 2.73,N 13.03。IR(KBr,cm-1):2 959(芳香族C—H),2 945(脂肪族 C—H),2 838(C—H),1 590(C=C),1 616(C—O),749(酞菁环)。1H NMR(600 MHz,DMSO):δ10.08~9.99(s,4H,aldehyde⁃H),8.26~8.23(m,1H,Ar⁃H),8.15(d,J=7.1 Hz,1H),8.10(dd,J=15.1 Hz,J=7.4 Hz,6H),7.99(dd,J=8.4 Hz,J=1.9 Hz,4H),7.94(dd,J=6.4 Hz,J=2.2 Hz,7H),7.88(t,J=3.2 Hz,4H),7.59(t,J=7.6 Hz,4H),7.55(dt,J=7.7 Hz,J=3.9 Hz,4H),7.50(dt,J=8.2 Hz,J=4.1 Hz,5H)。
Fe2Pc2(TA)4的制备:该制备过程与Zn2Pc2(TA)4相似,采用的金属盐为Fe(OAc)2·2H2O。产物为墨绿色粉末,产量0.260 0 g,产率52%,熔点大于300℃。元素分析按C46H24N8O6Fe计算的理论值(%):C 65.73,H 2.88,N 13.33。实验值(%):C 65.66,H 2.79,N 13.21。IR(KBr,cm-1):3 025(芳香族 C—H),2 870(脂肪族C—H),2 892(C—H),1 605(C=C),1 597(C—O),750(酞菁环)。
Co2Pc2(TA)4的制备:该制备过程与Zn2Pc2(TA)4相似,采用的金属盐为Co(OAc)2·2H2O。产物为深蓝色粉末,产量0.190 0 g,产率38%,熔点大于300℃。元素分析按C46H24N8O6Co计算的理论值(%):C 65.49,H 2.87,N 13.28。实验值(%):C 65.36,H 2.79,N 13.17。IR(KBr,cm-1):3 040(芳香族 C—H),2 930(脂肪族C—H),2 842(C—H),1 598(C=C),1 603(C—O),743(酞菁环)。
1.2.2 M2Pc2(TA)4/rGO复合材料的制备
将37.5 mg GO和110 mg M2Pc2(TA)4超声分散于2.5 mL DMF中,将得到的混合溶液转移至50 mL反应釜中,再加入20 mL DMF,鼓入氮气,100℃水热反应24 h。产物依次用DMF、蒸馏水和乙醇洗涤数次,得到复合材料M2Pc2(TA)4/rGO。
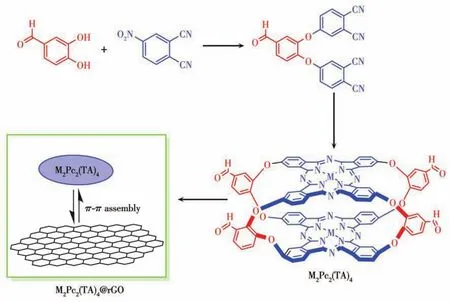
图1 M2Pc2(TA)4/rGO催化剂的制备示意图Fig.1 Preparation diagram of M2Pc2(TA)4/rGO catalyst
1.3 电催化活性测试
复合材料的ORR催化活性通过传统的三电极体系在电化学工作站上测试,以改性玻碳电极为工作电极,饱和甘汞电极为参比电极,铂丝电极为对电极,0.1 mol·L-1KOH溶液为电解液。测试时,使用湿润的镜头纸擦拭电极,取0.3 μm的α⁃氧化铝粉末于麂皮上,滴加少许蒸馏水,按照“8”字形路径对电极进行抛光。用去离子水冲洗电极表面,将电极头放在盛有少量去离子水和乙醇的烧杯中依次超声清洗2 min,用氮气吹干备用。测试时移取30 μL的2 mg·mL-1催化剂墨水均匀滴涂在玻碳电极(直径3 mm)上,红外灯下烘干。
CV测试在O2或N2饱和的0.1 mol·L-1KOH溶液中进行,扫描速率为5 mV·s-1,电位范围在-0.7~0.4 V(vs SCE)之间。LSV测试是在O2饱和的0.1 mol·L-1KOH溶液中进行,电极转速为1 600 r·min-1,扫描速率为5 mV·s-1,电位范围在-0.8~0.2 V(vs SCE)之间。
2 结果与讨论
2.1 M2Pc2(TA)4/rGO复合材料形貌分析
GO及Fe2Pc2(TA)4/rGO的SEM图及TEM图如图2所示。由图可知,GO表面褶皱不平,且彼此之间产生了随机的团聚现象,从而形成了无序的层状结构。如图2b所示,当负载Fe2Pc2(TA)4后,原GO结构特征消失,复合材料呈现出纳米团簇结构。GO的TEM图表明其表面呈褶皱状(图2c),球形金属酞菁很好地分散在rGO表面(图2d)。同时,可以看出金属酞菁负载之后,GO的层状结构部分被破坏。XRD表征结果进一步表明(图S1,Supporting informa⁃tion),M2Pc2(TA)4/rGO复合材料在8.5°处的GO特征衍射峰消失,在10.7°处出现新的衍射峰,说明GO在制备过程中被还原为rGO,其层状结构也被部分破坏[23]。

图2 (a)GO和(b)Fe2Pc2(TA)4/rGO复合材料的SEM图;(c)GO和(d)Fe2Pc2(TA)4/rGO复合材料的TEM图Fig.2 SEM images of(a)GO and(b)Fe2Pc2(TA)4/rGO composite;TEM images of(c)GO and(d)Fe2Pc2(TA)4/rGO composite
2.2 紫外可见光谱分析
图3为球形金属酞菁M2Pc2(TA)4、Zn2Pc2(TA)4/rGO以及GO的UV⁃Vis谱图。由图3a可知,M2Pc2(TA)4的谱图中出现了B带(或S带,290~350 nm)及Q带(600~700 nm)特征吸收峰。其中,Q带特征吸收峰的出现是由于酞菁环共轭结构中π电子从最高占据轨道(HOMO)跃迁到最低未占据轨道(LUMO),表明酞菁环生成[24]。从图3b还可以观察到GO在600~800 nm处没有明显的吸收峰,负载金属酞菁后,Zn2Pc2(TA)4的Q带吸收峰位置从660 nm红移至683 nm,表明复合材料之间的π⁃π作用减小了酞菁分子间的空间位阻效应,改变了酞菁环上电子云密度和空间分布[25],增大了π电子的流动,使得Zn2Pc2(TA)4的HOMO与LUMO轨道之间能带变窄,Q带吸收峰发生红移。
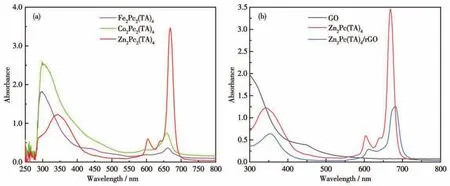
图3 (a)M2Pc2(TA)4及(b)Zn2Pc2(TA)4/rGO、Zn2Pc2(TA)4和GO的UV⁃Vis谱图Fig.3 UV⁃Vis spectra of(a)M2Pc2(TA)4,and(b)Zn2Pc2(TA)4/rGO,Zn2Pc2(TA)4,and GO
2.3 XPS分析
采用XPS进一步研究复合材料M2Pc2(TA)4/rGO的表面元素组成及化学状态。图4a为M2Pc2(TA)4/rGO及GO的XPS全谱图,从中可以看出组成GO的主要元素为C、O,与M2Pc2(TA)4复合后,除N元素外,谱图中分别显示Zn、Fe、Co元素的存在。图4b为Fe2Pc2(TA)4/rGO的Fe2p谱图,氧化态铁(Fe3+/Fe2+)的信号峰位于711.88 eV(Fe2p3/2)和725.28 eV(Fe2p1/2),说明Fe2Pc2(TA)4成功负载于rGO上[26]。GO的C1s谱图(图4c)在284.78、286.78 eV处分别出现了C—C/C=C和C—O的特征峰,复合材料Fe2Pc2(TA)4/rGO的C1s谱图中,C—N结合能出现在285.28 eV处,而且C—O键的结合能比GO低0.30 eV,表明在水热制备过程中GO被还原为rGO[27⁃28]。进一步分析N1s谱图(图4d)发现,Fe2Pc2(TA)4负载后,C—N与C=N的结合能变小,表明复合材料中rGO与金属酞菁之间存在强烈的π⁃π作用[29⁃30]。
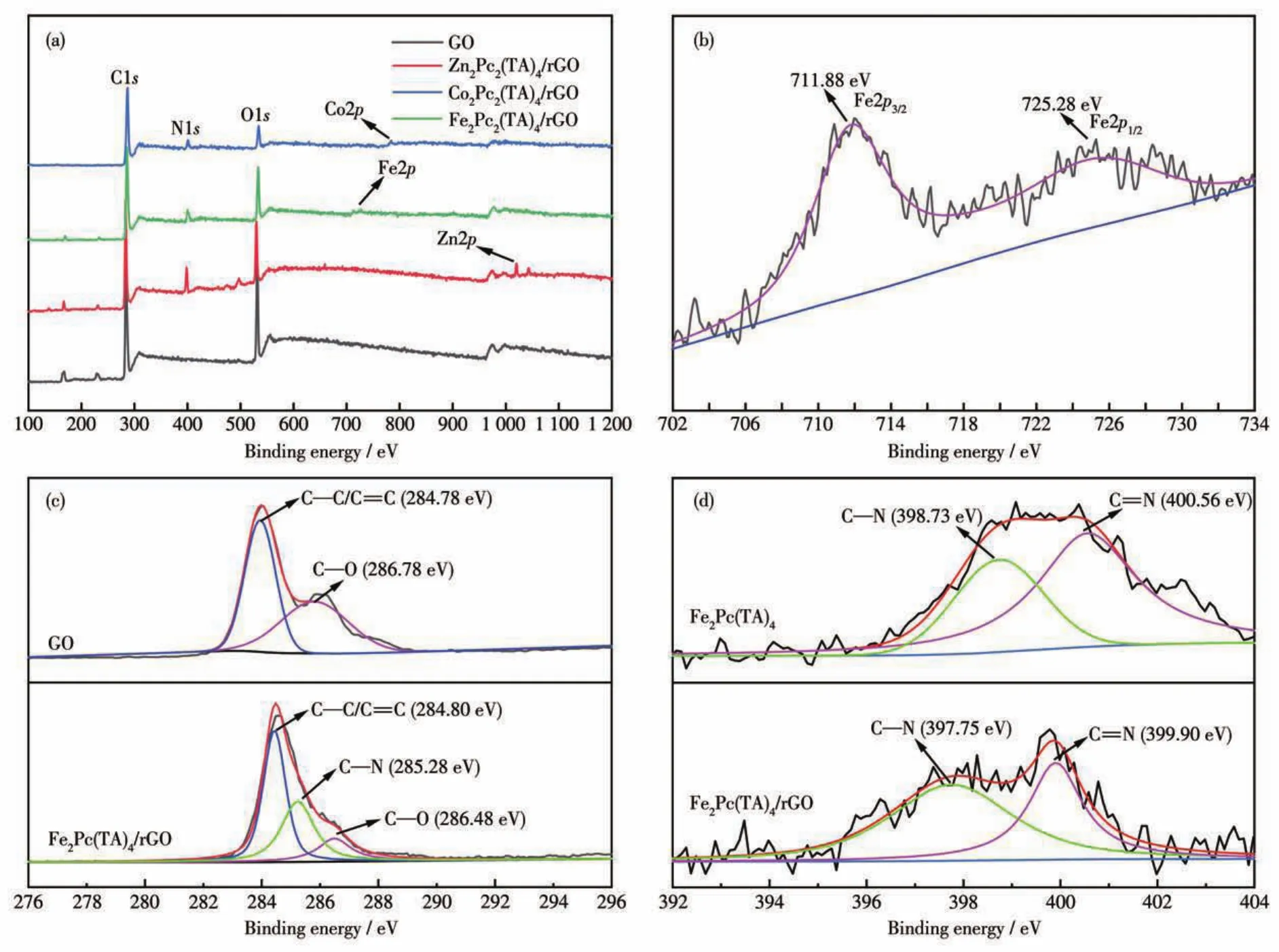
图4 (a)M2Pc2(TA)4/rGO和GO的XPS全谱图;(b)Fe2Pc2(TA)4/rGO的Fe2p谱图;(c)Fe2Pc2(TA)4/rGO和GO的C1s谱图;(d)Fe2Pc2(TA)4和Fe2Pc2(TA)4/rGO的N1s谱图Fig.4 (a)XPS survey spectra of M2Pc2(TA)4/rGO and GO;(b)Fe2p spectrum of Fe2Pc2(TA)4/rGO;(c)C1s spectra of Fe2Pc2(TA)4/rGO and GO;(d)N1s spectra of Fe2Pc2(TA)4and Fe2Pc2(TA)4/rGO
2.4 电化学测试
图5a为Fe2Pc2(TA)4/rGO和Fe2Pc2(TA)4在O2饱和的0.1 mol·L-1KOH中的CV曲线,由图可知,与Fe2Pc2(TA)4相比,Fe2Pc2(TA)4/rGO的氧还原峰电位较正,对应峰电流密度大,表明Fe2Pc2(TA)4/rGO复合材料具有更优异的ORR催化性能,rGO能有效提高Fe2Pc2(TA)4的催化活性。由图5b可知,Fe2Pc2(TA)4/rGO催化剂中Fe3+/Fe2+的氧还原峰电位(-0.11 V)比Pt/C催化剂的氧还原峰电位(-0.15 V)更正,表明Fe2Pc2(TA)4/rGO在0.1 mol·L-1KOH中表现出更高的氧还原催化活性。这是由于中心金属Fe2+在酞菁环形成的配位场中有利于其与氧配位,在氧化还原过程中更易被氧化为Fe3+[31],使ORR具有更低的过电势。进一步分析图5c、5d可知,Fe3+/Fe2+(-0.11 V)的氧还原电位比Zn3+/Zn2+(-0.13 V)和Co3+/Co2+(-0.16 V)的还原电位更高。结果表明,球形金属酞菁M2Pc2(TA)4中心金属离子M2+的d电子构型是影响M2Pc2(TA)4/rGO复合材料氧还原催化活性的重要因素。M2Pc2(TA)4/rGO催化ORR的活性顺序为Fe2Pc2(TA)4/rGO>Zn2Pc2(TA)4/rGO>Co2Pc2(TA)4/rGO。
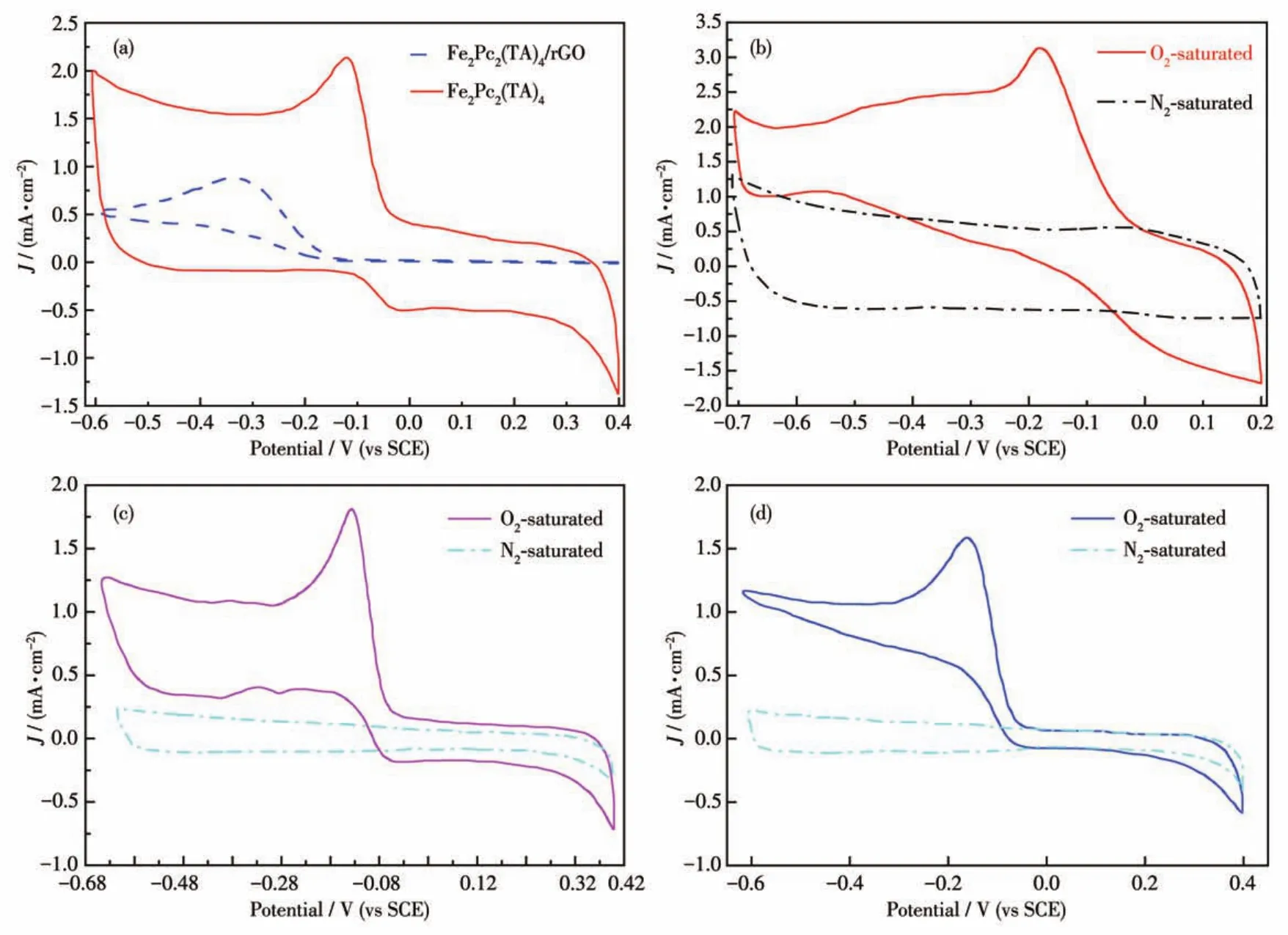
图5 (a)Fe2Pc2(TA)4/rGO、Fe2Pc2(TA)4的CV曲线;(b)Pt/C、(c)Zn2Pc2(TA)4/rGO、(d)Co2Pc2(TA)4/rGO在O2和N2饱和的0.1 mol·L-1KOH溶液中的CV曲线Fig.5 CV curves of(a)Fe2Pc2(TA)4/GO and Fe2Pc2(TA)4in O2⁃saturated 0.1 mol·L-1KOH solution;(b)Pt/C,(c)Zn2Pc2(TA)4/GO,and(d)Co2Pc2(TA)4/GO in O2⁃saturated and N2⁃saturated 0.1 mol·L-1KOH solution
在 O2饱和的 0.1 mol·L-1KOH 溶液和 0.1 mol·L-1KOH+3 mol·L-1CH3OH 溶液中,分别测试了Fe2Pc2(TA)4/rGO和Pt/C的CV曲线,评价其耐甲醇性能,扫速为100 mV·s-1,结果如图6所示。从图6a可以看出,Pt/C催化剂在-0.07 V出现了甲醇氧化峰,O2还原峰的电流密度比未加入甲醇时低,表明Pt/C催化剂的耐甲醇性能差;Fe2Pc2(TA)4/rGO复合材料在加入甲醇后未出现新的甲醇氧化峰,而且氧还原电位也并未发生明显变化,峰电位出现很小幅的负移(图6b)。上述结果表明Fe2Pc2(TA)4/rGO复合催化剂具有优异的耐甲醇性能,Fe2Pc2(TA)4/rGO有望替代Pt/C催化剂用于DMFC中。
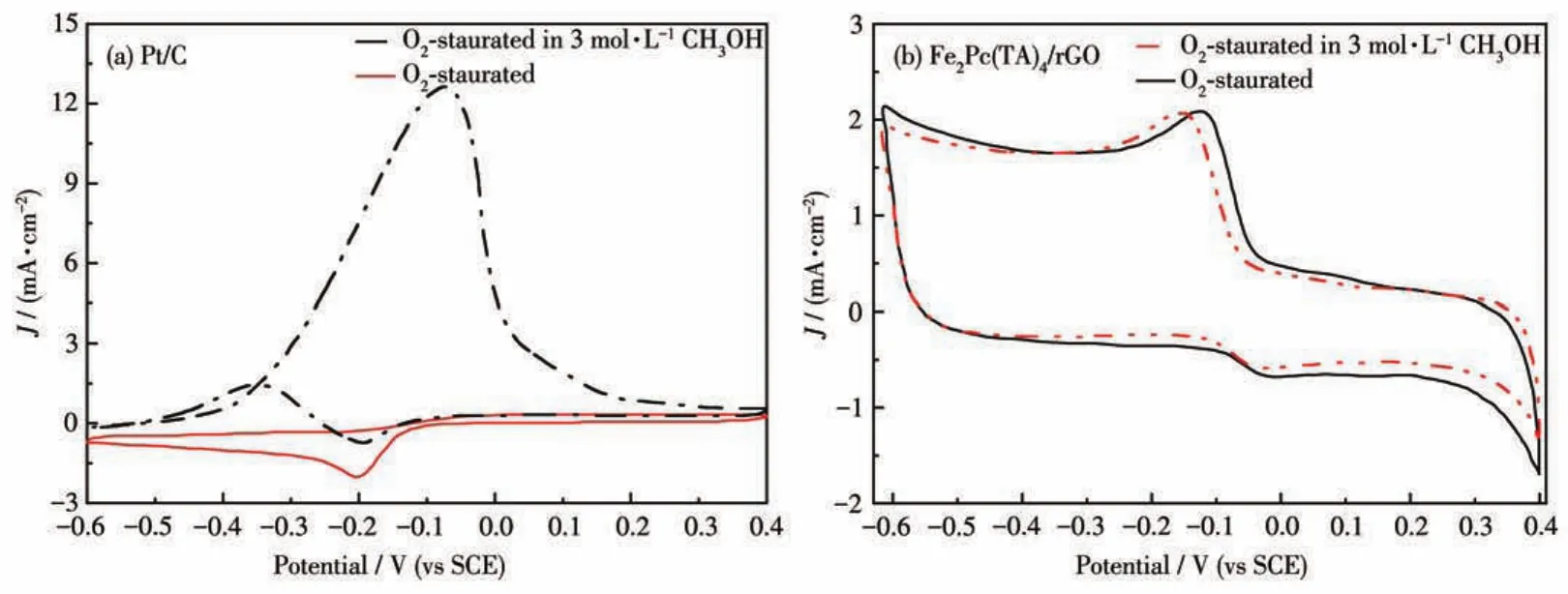
图6 (a)Pt/C和(b)Fe2Pc2(TA)4/rGO在O2饱和的0.1 mol·L-1KOH溶液中与含有3 mol·L-1CH3OH的0.1 mol·L-1KOH溶液中的CV曲线Fig.6 CV curves of(a)Pt/C and(b)Fe2Pc2(TA)4/rGO in O2⁃saturated 0.1 mol·L-1KOH solution and 0.1 mol·L-1KOH solution with 3 mol·L-1CH3OH
在O2饱和的0.1 mol·L-1KOH溶液中对Fe2Pc2(TA)4/rGO、Fe2Pc2(TA)4及商用Pt/C进行了LSV测试,进一步研究复合材料的ORR催化活性,结果如图7所示。图7a、7c、7e分别为不同转速下的LSV曲线,图7g为材料在1 600 r·min-1转速下的LSV曲线。由图可知,电流密度(J)逐渐增大并达到极限扩散电流密度(JL)。另外,Fe2Pc2(TA)4/rGO的起始电位-0.02 V 比 Pt/C(-0.07 V)和 Fe2Pc2(TA)4(-0.13 V)的起始电位分别减小50和110 mV,表明Fe2Pc2(TA)4/rGO催化剂在碱性介质中具有较好的氧还原催化性能。图7b、7d、7f分别为Fe2Pc2(TA)4/rGO、Fe2Pc2(TA)4、Pt/C的J-1与ω-1/2的关系图(ω是旋转圆盘的角速度),表明二者具有良好的线性关系,且不同电位下拟合的Koutecky⁃Levich(K⁃L)曲线斜率近似一致,表明ORR过程中转移电子数是相似的。
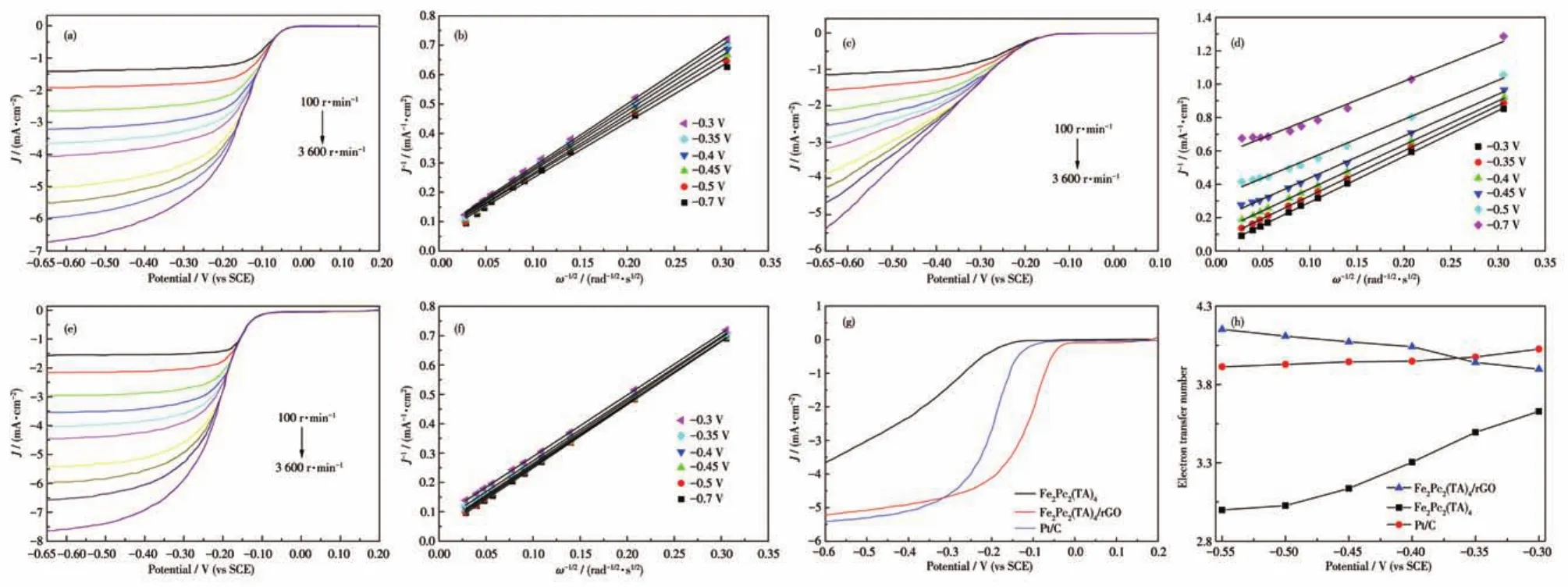
图7 (a)Fe2Pc2(TA)4/rGO、(c)Fe2Pc2(TA)4和(e)Pt/C在不同转速下O2饱和的0.1 mol·L-1KOH溶液中的LSV曲线;(b)Fe2Pc2(TA)4/rGO、(d)Fe2Pc2(TA)4和(f)Pt/C的K⁃L曲线;(g)Fe2Pc2(TA)4/rGO、Fe2Pc2(TA)4和Pt/C在转速为1 600 r·min-1下O2饱和的0.1 mol·L-1KOH 溶液中的LSV曲线;(h)Fe2Pc2(TA)4/rGO、Fe2Pc2(TA)4和Pt/C在-0.55~-0.3 V下的转移电子数Fig.7 LSV curves of(a)Fe2Pc2(TA)4/rGO,(c)Fe2Pc2(TA)4,and(e)Pt/C at different speeds in O2⁃saturated 0.1 mol·L-1KOH solution;K⁃L curves of(b)Fe2Pc2(TA)4/rGO,(d)Fe2Pc2(TA)4,and(f)Pt/C;(g)LSV curves of Fe2Pc2(TA)4/rGO,Fe2Pc2(TA)4,and Pt/C at 1 600 r·min-1in O2⁃saturated 0.1 mol·L-1KOH solution;(h)Electrons transfer number of Fe2Pc2(TA)4/rGO,Fe2Pc2(TA)4,and Pt/C at the voltage range from-0.55 to-0.3 V
ORR反应过程中的电子转移数n值可通过K⁃L方程计算获得,根据Koutecky⁃Levich定律,J与动力学电流密度(JK)和JL之间的关系如下[32]:
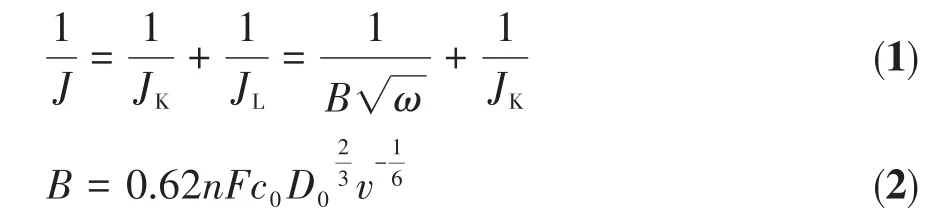
其中,F是法拉第常数(96 485 C‧mol-1),c0是O2的体积浓度(1.38×10-6mol·cm-3),D0是 O2的扩散系数(1.9×10-5cm2·s-1),v是电解质的运动黏度(0.01 cm2·s-1)。
根据K⁃L方程可计算在不同电位下的电子转移数n=3.89~4.15(图7h)。说明当用Fe2Pc2(TA)4/rGO作为催化剂催化ORR时遵循4电子反应过程(图8)。同时计算了Fe2Pc2(TA)4催化ORR时的电子转移数n=3.00~3.62,说明该还原过程也是4电子过程。研究表明,4电子反应过程可将氧分子还原成OH-,从电池能量角度来说,比2电子反应过程更有利,因而Fe2Pc2(TA)4/rGO具有更优异的催化活性。
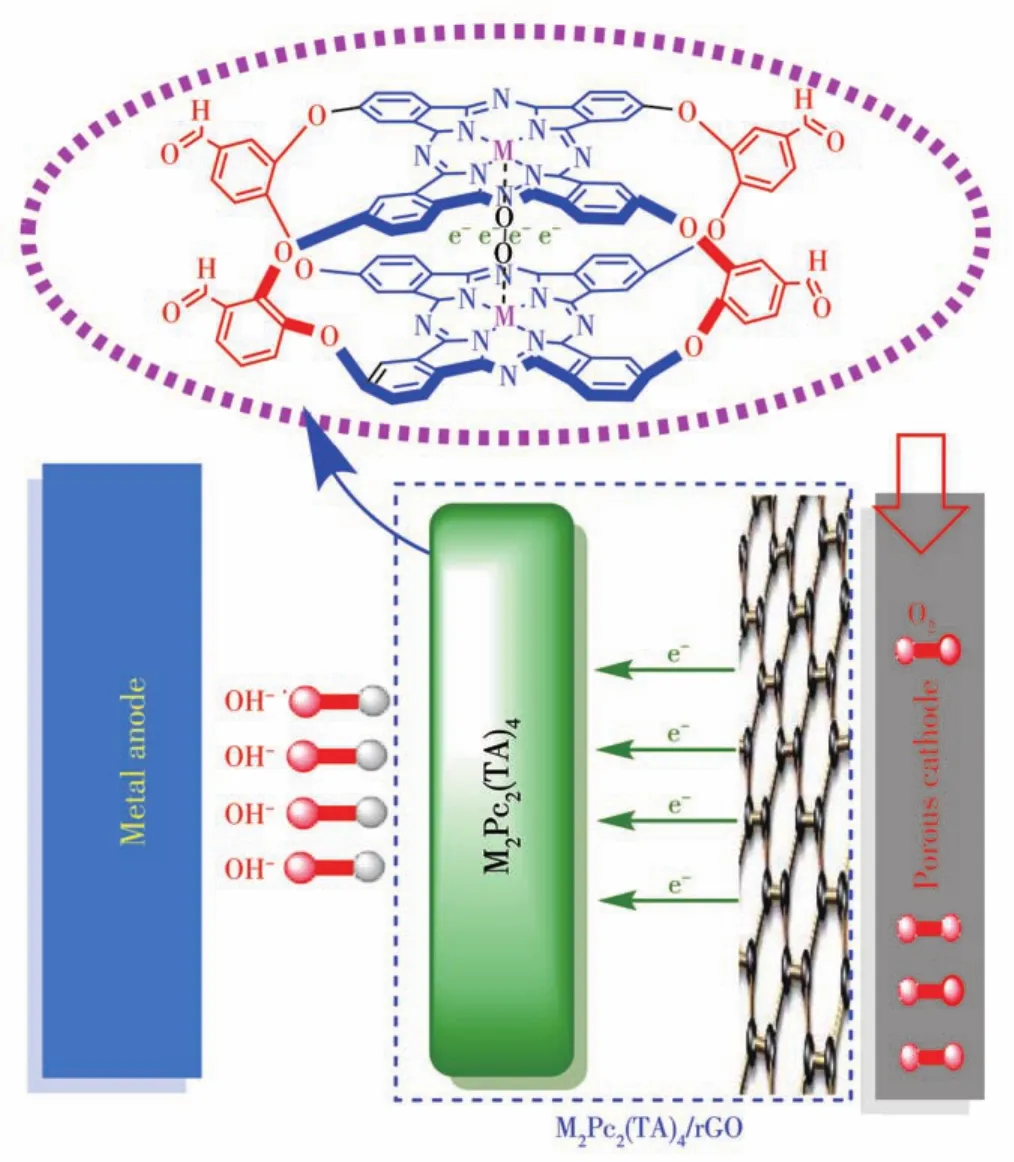
图8 M2Pc2(TA)4/rGO的催化机理图Fig.8 Diagram of the catalytic mechanism of M2Pc2(TA)4/rGO
3 结 论
基于金属酞菁具有仿生活化分子氧功能,通过水热技术将设计、合成的醛基取代球形金属酞菁M2Pc2(TA)4(M=Zn、Co、Fe)负载到GO上得到复合催化剂 M2Pc2(TA)4/rGO(M=Zn、Co、Fe)。形貌和结构表征结果表明球形金属酞菁通过“π⁃π堆积”作用负载在rGO表面后,酞菁的团聚现象得到明显改善;材料催化氧阴极反应活性评价结果表明rGO能与球形金属酞菁产生协同效应,促进ORR过程中电子的转移,有效提升电催化活性,ORR过程为4电子过程。Fe2Pc2(TA)4/rGO催化剂具有优异的耐甲醇性能。球形金属酞菁M2Pc2(TA)4的共轭结构、中心金属离子d电子构型和酞菁聚集状态是影响M2Pc2(TA)4/rGO复合材料氧还原催化活性的重要因素。不同中心金属离子的催化剂催化活性顺序为Fe2Pc2(TA)4/rGO>Zn2Pc2(TA)4/rGO>Co2Pc2(TA)4/rGO。本工作为DMFC中氧阴极非贵金属催化剂的研究提供了理论支持。
Supporting information is available at http://www.wjhxxb.cn
猜你喜欢
土壤学报(2022年3期)2022-08-27
全面腐蚀控制(2022年2期)2022-03-11
汽车实用技术(2022年4期)2022-03-07
中国听力语言康复科学杂志(2021年6期)2021-12-21
口腔护理用品工业(2021年4期)2021-11-02
安徽工业大学学报(自然科学版)(2021年3期)2021-09-08
上海节能(2021年1期)2021-02-04
装备维修技术(2020年5期)2020-11-20
分析化学(2018年1期)2018-01-18
分析化学(2017年9期)2017-10-16
