
Slc4a11基因缺失在CHED发病中的作用
2022-01-14 12:45:54邱颖萍牛国桢刘春雨邵玉婷毕燕龙
同济大学学报(医学版) 2021年6期
邱颖萍,牛国桢,刘春雨,邵玉婷,毕燕龙
(同济大学附属同济医院眼科,上海 200065)
先天性遗传性角膜内皮营养不良(congenital hereditary endothelial dystrophy,CHED)是一种罕见的角膜内皮遗传性疾病,以双侧弥漫性角膜水肿和混浊为特征,可在生命早期出现不同程度的视力损伤[1]。目前角膜移植术是其唯一有效的治疗方法[2-3],但全球角膜供体的紧缺、手术并发症和手术技术难度大等限制了该方法的普及,寻求非侵入性的治疗干预新靶点具有重要意义[4]。SLC4A11作为碳酸氢盐转运蛋白家族的一种跨膜蛋白[5],其突变可导致CHED和Fuchs角膜内皮营养不良(Fuchs’ endoth-elial corneal dystrophy, FECD)的发生[6-8]。SLC-4A11作为一种分子泵作用于角膜内皮,可以促进水的跨膜转运、Na+/OH-共转运、Na+非依赖的H+(OH-)转运和NH3的转运[9-10],对防止角膜水肿,维持角膜稳态起到重要作用。研究表明SLC4A11缺失与氧化损伤和凋亡的敏感性增加有关[11-12]。自噬是细胞在压力条件下通过降解胞浆成分维持细胞稳态的一种高度保守的细胞机制。病理条件下,自噬功能受损将会导致线粒体受损,增强细胞氧化应激损伤,参与角膜营养不良等多种疾病的发病过程[13]。目前有关SLC4A11的具体生理作用及其在角膜内皮营养不良中的作用机制尚不清楚,成为近年来的研究热点。
本研究利用CRISPR/Cas9技术敲除C57BL/6J小鼠Slc4a11基因,观察基因敲除后小鼠角膜的变化,同时探究Slc4a11基因缺失在CHED发病过程中对自噬的潜在调控作用机制,以期为角膜内皮营养不良性疾病的发病机制和防治方法提供新的研究思路和干预靶点。
1 材料与方法
1.1 试剂与设备
质粒px330-Cas9购自Addgene公司;MEGAsh-ortscriptTMT7 Transcription Kit、mMESSAGE mM-ACHINE T7 Kit购自美国Life Technologies公司;TRIeasyTMTotal RNA Extraction Reagent、Hieff®qPCR SYBR Green Master Mix购自上海翊圣生物科技有限公司;SLC4A11抗体购自美国Signalway Antibody公司;鼠尾基因型快速鉴定试剂盒、辣根过氧化物酶标记山羊抗兔IgG(H+L)购自上海碧云天生物技术有限公司;β-actin抗体购自美国Proteintech公司;LC3抗体、p62抗体、Beclin1抗体购自英国Abcam公司;显微镜型号购自德国Leica公司。
1.2 转基因小鼠的构建和繁殖
针对C57BL/6J小鼠Slc4a11基因内含子8和内含子13设计2条sgRNA,sgRNA1:5′-CAGCAT-TTGCCAGGGATGCA-3′和sgRNA2:5′-CTGTTG-CAGTGAGCAAGTTC-3′。根据sgRNA结构设计sgRNA PCR扩增上游引物Slc4a11sgRNA1,Slc4a11sgRNA2,下游引物T7SG-LW,见表1。利用MEGA shortscriptTMT7 Transcription Kit体外转录sgRNA。以px330-Cas9为模板,利用引物T7Cas9-F和T7Cas9-R进行PCR扩增,见表1。利用mMESSAGE mMA-CHINE T7 kit体外转录Cas9 mRNA。将转录好的sgRNA和Cas9 mRNA混合并调整浓度至50 ng/μL,显微注射法注入C57BL/6J小鼠(购自上海杰思捷实验动物有限公司)受精卵胞质中,再植入假孕小鼠体内,繁殖得到F0代小鼠。将基因敲除阳性的F0代小鼠经过两代繁育后获得F2代小鼠。选取F2代中指定年龄的基因敲除纯合子小鼠(knockout, KO)和C57BL/6J野生型小鼠(wild type, WT)作为实验对象,用于后续实验。
1.3 基因型鉴定
剪取0.2~1 cm的小鼠尾尖,使用鼠尾基因型快速鉴定试剂盒提取基因组DNA,作为PCR模板。鉴定所需PCR扩增引物包括突变型鉴定引物Slc4a11Outer check 2F、Slc4a11Outer check 2R,野生型鉴定引物Slc4a11-del-check-F、Slc4a11-del-check-R,见表1。利用PCR纯化试剂盒纯化PCR产物,取5 μL进行1.5%琼脂糖凝胶电泳,部分产物进行DNA测序。

表1 引物名称及其序列
1.4 角膜内皮茜素红染色
取20周小鼠,WT组和KO组各3只。在显微镜下剪取小鼠角膜组织,内皮面朝上放置于载玻片上,0.2%茜素红染液(pH 4.2)染色90 s,1×PBS洗涤3次。4%多聚甲醛固定10 min,洗涤后盖上盖玻片,显微镜拍照观察。每个角膜在显微镜下随机选取5个不重复的0.1 mm×0.1 mm正方形区域,用Image J图像处理软件评估CECs的密度和面积。
1.5 眼前节光学相干断层扫描(anterior segment optical coherence tomography, AS-OCT)
取10、20、30周小鼠,每个年龄段的WT组小鼠和KO组小鼠各3只。所有操作均由同1名有经验的技师完成。应用德国Zeiss公司AS-OCT对小鼠进行眼前节的照相,测量中央角膜厚度。每只眼测3次,取平均值。
1.6 角膜组织切片H-E染色
取10周的WT和KO小鼠眼球各3只,浸于10%酸性甲醛中固定48 h,常规脱水,将眼球沿矢状位方向剖为3个部分,取中间部分进行乙醇逐级脱水、透明、浸蜡,石蜡包埋后常规切片,行H-E染色。在光学显微镜下观察角膜组织形态、采集图像,利用Case Viewer 2.2软件测量角膜中央厚度,每张图像测3次,取平均值。
1.7 RT-qPCR及Western印迹法检测
取10、30周的WT和KO小鼠各3只,分离出小鼠角膜的后弹力层和角膜内皮,TRIzol法提取总RNA,反转录获得cDNA,进行real-time PCR,以β-actin为内参,采用2-ΔΔCt方法计算各基因相对表达量。引物序列见表2。PCR反应体系20 μL,反应条件为95 ℃变性10 s,60 ℃复性20 s,72 ℃延伸20 s,共40个循环。

表2 RT-qPCR的引物名称及其序列
取10、30周的WT和KO小鼠各3只,将分离出的后弹力层和角膜内皮组织匀浆后,提取并测定总蛋白浓度。各样本等质量等体积上样,80 V恒压电泳。200 mA湿转120 min,无蛋白快速封闭液封闭10 min,TBST洗膜3次,加入抗Slc4a11、LC3、Beclin1、p62、β-actin(1∶1 000)等一抗,4 ℃孵育过夜,洗膜,二抗室温孵育2 h,洗膜,加入显影剂,于凝胶成像系统曝光检测蛋白表达。
1.8 透射电子显微镜
将各组角膜样本置于2.5%的戊二醛溶液,4 ℃固定过夜,再于1%锇酸中进行二次固定。经漂洗后进行梯度乙醇逐级脱水。将角膜浸入环氧树脂进行包埋,之后进行半薄切片,切片厚度为1 μm,天青蓝染色后光镜下观察。然后进行超薄切片,厚度为80 nm,收集于铜网上。用柠檬酸铅染色液染色15 min,透射电镜下观察拍照。
1.9 统计学处理

2 结 果
2.1 Slc4a11基因敲除小鼠的验证
应用CRISPR-Cas9技术敲除Slc4a11基因外显子9-13,获得Slc4a11基因敲除小鼠。部分F2代小鼠基因型鉴定结果见图1,包含Slc4a11+/-(出现589 bp突变型条带和469 bp野生型条带)、Slc4a11+/+(只出现469 bp野生型条带)和Slc4a11-/-(只出现589 bp突变型条带)3种基因型。Slc4a11-/-小鼠的DNA测序结果显示有2 100 bp碱基的缺失,外显子9-13被敲除,见图1。在Slc4a11-/-小鼠角膜内皮组织中未检测到SLC4A11蛋白和mRNA的表达,见图2,表明小鼠角膜内皮组织中Slc4a11基因被成功敲除。
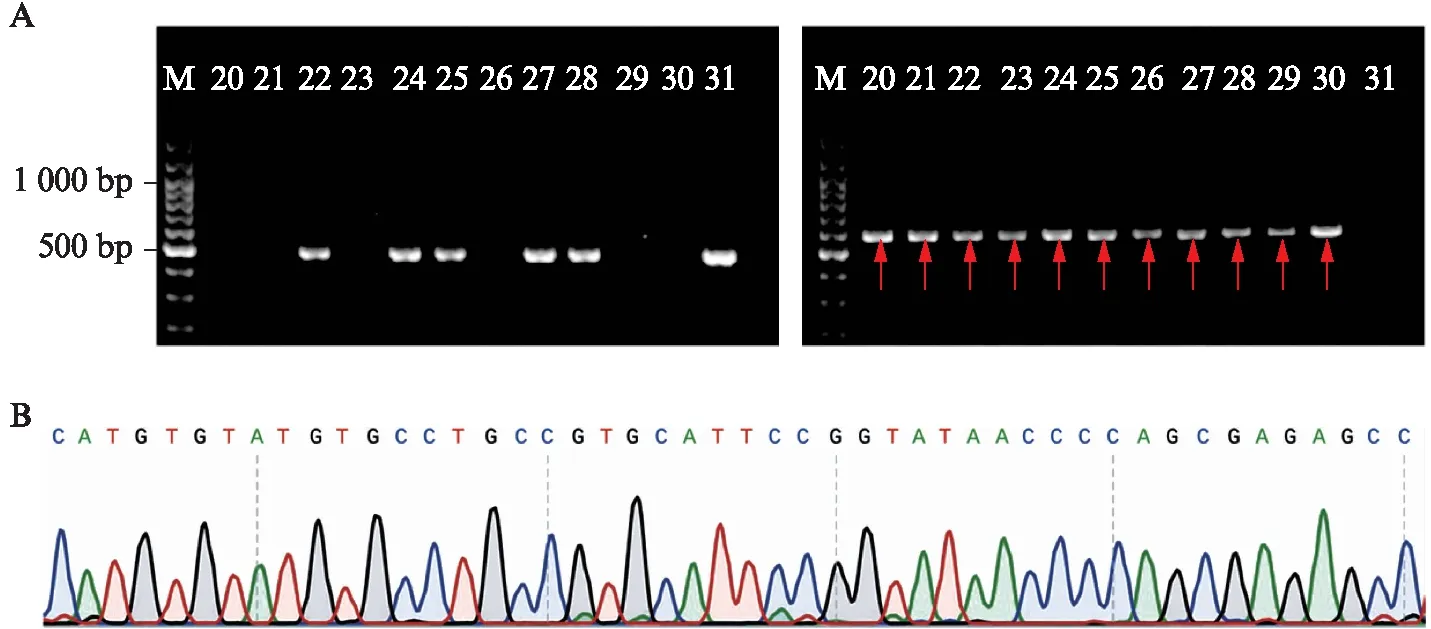
图1 Slc4a11敲除小鼠F2代仔鼠20~31号基因型鉴定结果及DNA测序峰图
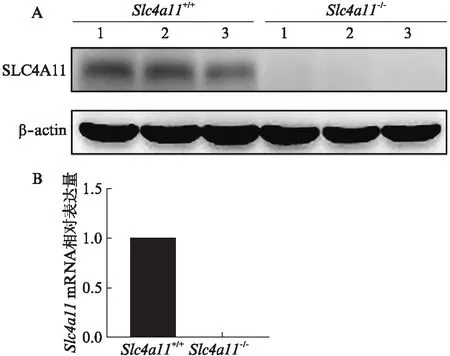
图2 Slc4a11+/+和Slc4a11-/-小鼠角膜内皮中SLC4A11蛋白(A)和mRNA(B)的表达
2.2 Slc4a11基因敲除对小鼠角膜表型的改变
2.2.1 角膜内皮茜素红染色 20周的KO小鼠CECs密度明显小于相同年龄的WT小鼠[(2 767±63)vs(3 252±193)cells/mm2],差异有统计学意义(P<0.05);同时CECs面积明显大于WT小鼠[(242.7±5.0)vs(188.0±13.5)μm2,]差异有统计学意义(P<0.05),见图3。

图3 WT和KO小鼠角膜内皮细胞茜素红染色
2.2.2 AS-OCT AS-OCT结果显示WT组小鼠角膜厚度随着年龄的增长呈相对稳定的趋势,而KO组小鼠角膜厚度随着年龄的增长而进行性增厚;同时相同年龄的KO小鼠角膜厚度要明显大于WT小鼠[10周:(100.0±10.0)vs(136.7±5.8)μm;20周:(100.0±10.0)vs(163.3±5.8)μm,30周:(103.3±5.8)vs(176.7±5.8)μm],差异均有统计学意义(P<0.05),见图4。
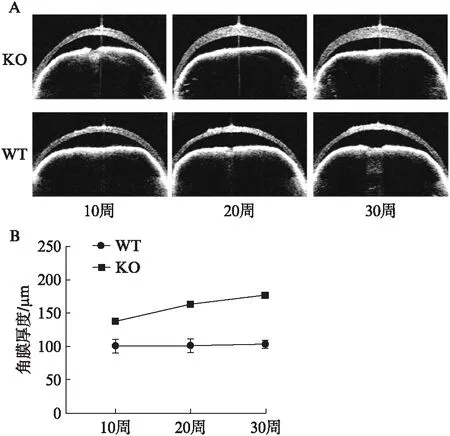
图4 WT和KO小鼠的AS-OCT检测结果
2.2.3 H-E染色和透射电镜 H-E染色显示WT小鼠角膜组织结构完整,各层结构清晰,基质层纤维排列规整,CECs呈单层扁平状,无明显水肿。相较于WT小鼠,KO小鼠角膜厚度明显增加[(84.4±5.8)vs(127.4±13.4)μm]差异有统计学意义(P<0.05),同时可见基质层水肿,厚度增加,基质层纤维排列欠规则,纤维裂隙增多;以及CECs明显水肿,出现大量空泡样改变,见图5。透射电镜同样显示KO小鼠出现CHED特征的角膜病理改变,包括:角膜后弹力层增厚;CECs增大;CECs出现空泡化改变等,见图6。
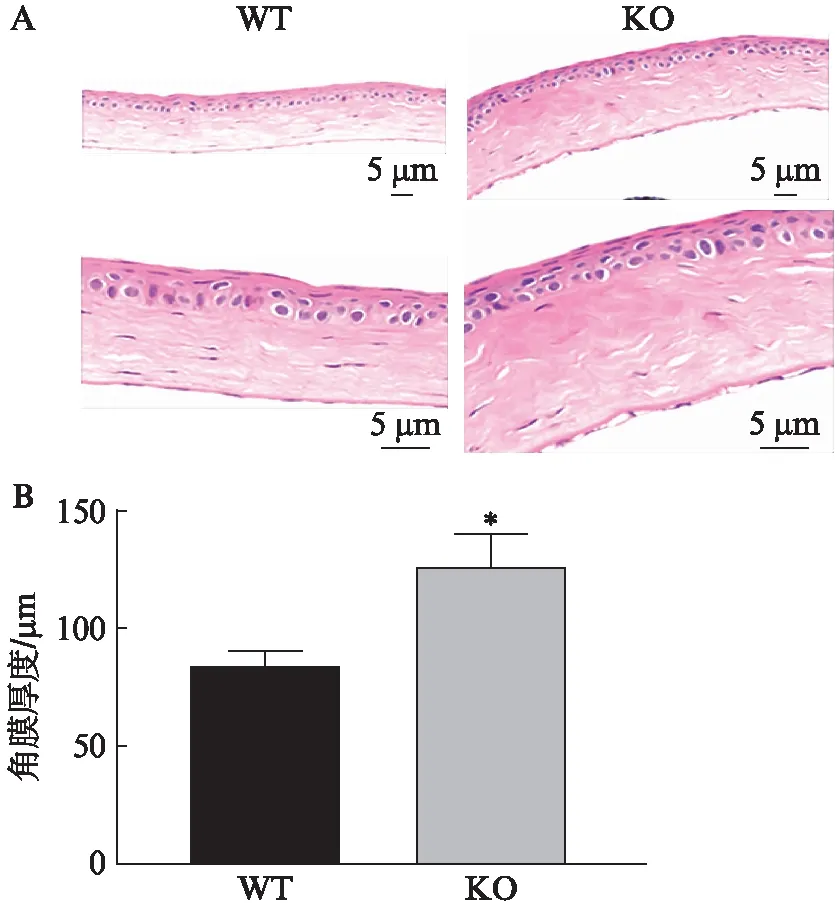
图5 10周龄的WT和KO小鼠角膜组织H-E染色
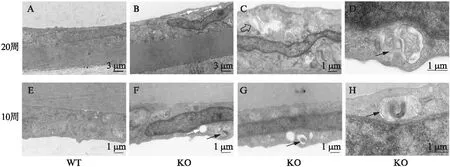
图6 10周龄和20周龄WT和KO小鼠角膜组织透射电镜图像
2.3 Slc4a11基因敲除对小鼠角膜内皮自噬信号通路的影响
RT-qPCR检测自噬信号通路相关因子显示,在10周和30周的小鼠角膜内皮中,相较于同年龄WT组,KO组自噬相关因子LC3、Beclin1的表达明显升高,p62表达降低,差异有统计学意义(P<0.05),见图7。
Western印迹法检测同样显示,在10周和30周的小鼠角膜内皮中,相较于同年龄WT组,KO组自噬相关蛋白LC3 Ⅱ/Ⅰ、Beclin1的表达明显升高,p62表达降低,差异有统计学意义(P<0.05),见图7。
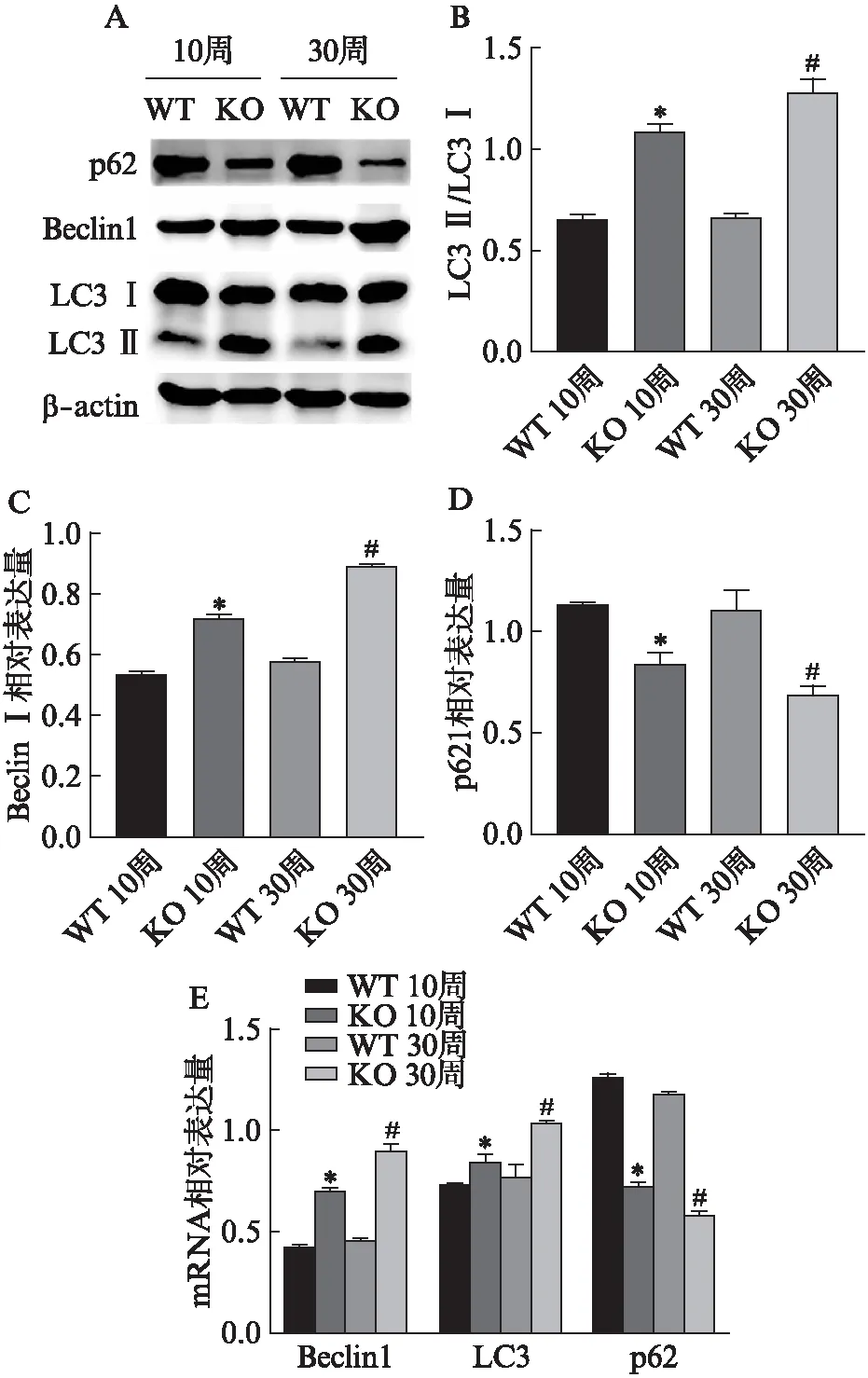
图7 Slc4a11敲除对小鼠角膜内皮自噬相关因子表达水平的影响
透射电镜显示10、20周的KO小鼠CECs内均可见自噬体及其吞噬的内容物,而WT小鼠CECs内很少能观察到自噬体结构,见图6。
3 讨 论
2006年,Vithana等[14]指出位于染色体20p13区域的Slc4a11基因突变导致了CHED的发生。Slc4a11突变通过多种方式引起CHED的发病。Slc4a11突变造成SLC4A11蛋白分子错误折叠,使其滞留于内质网,无法到达质膜实现转运功能,抑或突变蛋白虽可以到达细胞表面,但未携带某些功能结构,仍无法实现转运功能[4,15]。目前有关SLC4A11在CHED发病过程中的具体作用机制尚不清楚。本研究利用CRISPR/Cas9技术敲除小鼠Slc4a11基因,在动物模型上探究SLC4A11在角膜内皮营养不良发病过程中的作用及相关机制。
本研究利用CRISPR/Cas9技术敲除小鼠Slc4a11基因外显子9-13,结果显示KO小鼠显现出与CHED相似的表型特征,包括角膜厚度增厚、基质层水肿和胶原纤维排列紊乱、后弹力层增厚、CECs增大和空泡化改变。本研究中20周的KO小鼠的CECs密度相较于WT小鼠明显减小,同时CECs面积明显增大。这说明CECs损伤和凋亡的出现早于Han等[16]报导的40周。这是由于Slc4a11缺失激活凋亡通路,从而抑制角膜内皮细胞生长并降低其活力[17-18]。近来有研究指出,SLC4A11作为一种细胞黏附分子,增强了角膜内皮细胞对后弹力层的黏附作用,因此,Slc4a11的缺失会减弱这种黏附作用,使角膜内皮细胞数量减少[19]。
Slc4a11基因缺失通过诱导小鼠角膜的病理改变成功模拟出与CHED相似的疾病表征,在此基础上,本研究进一步探究Slc4a11缺失在CHED发病中的潜在致病机制。FECD患者角膜内皮组织中,线粒体自噬水平的上调导致功能性线粒体数量的减少。降低自噬流水平可能对FECD患者具有治疗作用[14]。Choi等[13]指出自噬功能缺陷可能在颗粒状角膜营养不良2型的发病机制中发挥重要作用。可以看出,自噬活动在相关的角膜病变发挥着重要作用[20]。因此,Slc4a11基因缺失是否通过影响机体的自噬活动导致疾病的发生。本研究结果显示,相较于同年龄的WT小鼠,KO小鼠角膜内皮中的自噬相关因子LC3、Beclin1表达上升,而p62表达下降。以上结果表明Slc4a11的敲除使小鼠角膜内皮组织的自噬水平显著升高。Slc4a11基因缺失可能通过增强机体自噬水平诱导CHED的发生发展。Guha等[21]发现在Slc4a11敲除的人角膜内皮细胞模型中,在氧化应激存在的情况下,细胞内ROS的生成增多,角膜内皮细胞活力减低。同时,Slc4a11敲除的角膜内皮细胞和CHED患者的角膜内皮组织都出现Nrf2及Nrf2下游抗氧化相关蛋白表达的下降。这些结果表明Nrf2信号通路及氧化应激同样参与了CHED的发病过程。而自噬通过自噬特异底物p62又与Nrf2信号通路密切联系,p62的累积可以激活Nrf2信号通路,上调下游抗氧化相关蛋白的表达,在角膜和视网膜等病变中发挥重要作用。有研究报导角膜缘干细胞可以通过自噬-Nrf2-ARE信号通路更加严格地调控细胞氧化还原反应[22]。同时有研究指出FECD患者角膜内皮中DJ-1和Nrf2相关抗氧化蛋白表达量的下降可能与自噬水平的改变有关[23]。由此推测,在CHED的发病过程中,Slc4a11的缺失导致自噬水平上调,引起自噬降解底物p62水平的下降,减弱了由p62介导的对Nrf2信号通路的激活作用,从而使抗氧化相关蛋白表达下降,细胞应对氧化应激能力减弱,导致细胞的损伤与凋亡。以上有关Slc4a11与自噬以及Nrf2信号通路在角膜内皮营养不良中的具体作用关系,本课题组将进一步研究和证实。
综上所述,本研究利用CRISPR/Cas9技术构建了Slc4a11基因敲除小鼠模型[24],并证实该基因敲除小鼠模型具有与CHED相似的表型特征,为今后将其作为探究Slc4a11在CHED发病中的作用机制及治疗策略的实验工具提供了理论上的支持。同时,本研究发现在Slc4a11基因敲除小鼠的角膜内皮组织中存在自噬水平升高的现象,Slc4a11与自噬以及Nrf2信号通路可能在CHED的发病过程中共同发挥调控作用,靶向这些过程可能可以为治疗CHED提供新的研究思路和靶点。
猜你喜欢
科教新报(2023年38期)2023-10-05 19:22:53
中国医药指南(2017年3期)2017-11-13 02:55:28
华东师范大学学报(自然科学版)(2017年1期)2017-02-27 13:41:06
高师理科学刊(2016年8期)2016-06-15 20:27:48
中国组织化学与细胞化学杂志(2016年4期)2016-02-27 11:16:04
安徽医科大学学报(2015年9期)2015-12-16 11:09:42
医学研究杂志(2015年8期)2015-06-22 14:00:57
医学研究杂志(2015年12期)2015-06-10 06:57:46
医学研究杂志(2015年11期)2015-06-10 06:44:03
山西大同大学学报(自然科学版)(2015年1期)2015-01-22 07:14:05
