
过渡金属改性的Mg-Al复合氧化物催化尿素与苯酚绿色合成水杨酰胺
2022-01-13 00:25张学兰魏淑伟吴鸿伟赵雪英肖瑞瑞王振华路文学王登峰
石油学报(石油加工) 2022年1期
张学兰,张 涛,魏淑伟,吴鸿伟,赵雪英,肖瑞瑞,王振华,路文学,陈 政,王登峰
(1.枣庄学院 化学化工与材料科学学院,山东 枣庄 277160;2.山东中实易通集团有限公司,山东 济南 250002;3.兖矿水煤浆气化及煤化工国家工程研究中心有限公司,山东 济南 250101)
水杨酰胺(SA),又称2-羟基苯甲酰胺,可作为医药、农药、香料、橡胶及有机合成绿色中间体,同时它也是治疗头痛、神经痛、关节痛及活动性风湿症等疾病药品[1]。传统水杨酰胺生产路线是苯酚与NaOH反应生成苯酚钠,然后苯酚钠与CO2作用,经硫酸化得到水杨酸,最后氨化获得水杨酰胺[2]。上述合成路线工艺流程长、设备投入大、生产成本高、酸碱废液排放量大。而从尿素和苯酚出发合成水杨酰胺技术具有原料便宜、工艺简单等优点,且所副产的氨气可与CO2耦合生成尿素,实现了CO2间接合成水杨酰胺[3],符合绿色化工和可持续发展的要求,从而引起广泛关注。2008年,孙予罕课题组[4]报道ZnO可催化尿素和苯酚合成水杨酰胺,但其易与尿素热分解产物作用生成可溶的Zn(NCO)2(NH3)2,因此其只是该工艺均相催化剂前驱体。2013年,笔者借助碱金属修饰的MgO催化剂,实现了多相催化水杨酰胺的合成[5]。然而,上述催化剂比表面积低(<10 m2/g)、表面碱强度不高(<130 μmol/g),水杨酰胺收率有待提高。同时,尚未见其他课题组对尿素和苯酚合成水杨酰胺催化剂的相关研究报道。因此,急需通过一定方法获得高活性合成水杨酰胺的多相固体催化剂。
除催化剂组成外,前驱体结构对催化剂物化性质和催化性能有较大影响。Mg-Al水滑石(Mg6Al2(OH)16CO3·nH2O)是一种层状化合物,具有“记忆效应”:将其在低于600 ℃温度下煅烧,使Mg-Al水滑石层状结构发生塌陷,在失去层间水分子和阴离子的同时,获得Mg-Al复合氧化物。然后,将上述复合氧化物于金属(Mx+,x为2或3)硝酸盐水溶液中搅拌,借助水中CO32-,可重新获得具有层状结构的Mg-Al-M类水滑石[6];再将Mg-Al-M在适宜温度下热处理,即可得到Mx+修饰的Mg-Al复合氧化物。上述催化材料具有比表面积大、金属离子分散性好、碱性强等优点,被广泛用于碱催化反应[7-10]。最近,笔者所在课题组分别借助Mg-Al-La复合氧化物[10]、Mg-Al-Y复合氧化物[11]和Mg-Al-Zr复合氧化物[12]实现了尿素醇解多相催化合成有机碳酸酯。考虑到苯酚与醇分子都具有羟基活泼氢原子,所以,尿素和苯酚反应与尿素醇解反应具有相似性。因而,作为研究工作的延续,有必要考察金属离子修饰的Mg-Al复合氧化物催化合成水杨酰胺反应性能。
本研究中,借助水滑石“记忆效应”,笔者将过渡金属离子引入Mg-Al水滑石层板,煅烧得到金属离子修饰的Mg-Al复合氧化物,显著提升了催化剂碱性。并考察了它们催化合成水杨酰胺反应性能,基于催化剂表征和反应测试,建立了催化剂表面碱性质与催化性能之间的内在联系。最终,依据催化剂重复性实验,获得了催化活性高、催化稳定性好、制备简单的多相合成水杨酰胺的固体碱催化剂。
1 实验部分
1.1 原料和试剂
尿素、Mg(NO3)2·6H2O、Al(NO3)3·9H2O、Cr(NO3)3·9H2O、Zn(NO3)2·6H2O、Co(NO3)2·6H2O、Cu(NO3)2·3H2O、Fe(NO3)3·9H2O、Zn(NO3)2·6H2O和水杨酰胺(SA),国药集团化学试剂有限公司产品;NaOH、Na2CO3、苯酚,阿拉丁试剂(上海)有限公司产品;4-羟基苯甲酰胺(HA)、呫吨酮(XA),百灵威(北京)科技有限公司产品;2,4,6-三(2-羟苯基)-1,3,5-均三嗪(HST),西格玛奥德里奇中国有限公司产品。本实验中所使用试剂均为分析纯。
1.2 催化剂制备
采用共沉淀法制备Mg-Al水滑石(HT-0),制备流程参照文献[7]:剧烈搅拌下,将分别含有Mg(NO3)2·6H2O和Al(NO3)3·9H2O(Mg2+和Al3+起始摩尔比为3∶1)、NaOH和Na2CO3的50 mL 水溶液同时滴入50 mL去离子水中,保持pH=10。滴定完成后,常温陈化6 h,减压抽滤和去离子水洗涤至滤液中性,100 ℃烘干10 h,获得HT-0样品。
将HT-0样品在450 ℃、N2中焙烧5 h,获得Mg-Al复合氧化物(CHT-0)。
借助水滑石“记忆效应”,制备过渡金属改性的Mg-Al-M类水滑石(HT-M,M为Cr3+、Mn2+、Fe3+、Co2+、Cu2+):称取1.0 g CHT-0样品浸渍于100 mL、0.1 mol/L的M(NO3)x水溶液中,常温搅拌48 h、悬浊液减压抽滤、去离子水洗涤数次后,100 ℃烘干10 h,得到HT-M样品。
将HT-M样品在450 ℃、N2中焙烧5 h,制得过渡金属改性的Mg-Al复合氧化物(CHT-M)样品。
1.3 催化剂样品表征
采用日本理学D/max-rA型X射线衍射仪进行样品的X射线衍射(XRD)分析(CuKα辐射,Ni滤光片),管电压40 kV,管电流40 mA,扫描范围5°~75°,扫描速率5 °/min。采用美国Micromeritics公司ASAP2020型自动物理吸附仪测试催化剂样品的吸附-脱附曲线,测试前对样品进行300 ℃真空脱气6 h,采用BET方程计算比表面积,通过BJH模型分析孔径。采用美国FET公司XL30 S-FEG型扫描电子显微镜(SEM)分析催化剂样品形貌,加速电压10 kV。采用美国TJA公司AtomScan16型电感耦合等离子体原子发射光谱仪(ICP-AES)测定催化剂样品金属组成。采用自制装置进行CO2程序升温脱附(CO2-TPD)实验:将200 mg催化剂放入石英反应管中,在N2中400 ℃下处理3 h,冷却至25 ℃吸附CO2至饱和,再用N2吹扫2 h;然后以升温速率10 ℃/min升温至600 ℃,采用瑞士Balzer公司Omnistar-2000型质谱仪采集信号。
1.4 催化性能评价
催化剂催化性能评价典型过程如下:将0.3 g新鲜催化剂、23.5 g苯酚、3 g尿素加入100 mL带有0.7 m冷凝回流管和磁力搅拌不锈钢反应釜中,待温度升至210 ℃,打开磁力搅拌器搅拌,至反应结束(12 h);反应器冷却到25 ℃,将反应混合物离心分离除去催化剂。采用日本岛津公司LC-2010ATvp型高效液相色谱(VPODS-18不锈钢色谱柱、流动相甲醇)测定液体产物的组成。借助水杨酰胺收率(ySA,%)、水杨酰胺选择性(sSA,%)、4-羟基苯甲酰胺选择性(sHA,%)、呫吨酮选择性(sXA,%)和2,4,6-三(2-羟苯基)-1,3,5-均三嗪选择性(sHST,%)评价催化剂催化性能。
(1)
(2)
(3)
(4)
(5)
式(1)~式(5)中:nurea,0为反应开始时反应体系中尿素物质的量,mol;nSA、nHA、nXA和nHST分别为反应结束后液态产物中SA、HA、XA和HST物质的量,mol。
1.5 重复性实验
以CHT-Mn为模板催化剂考察催化剂重复使用性。反应结束后,反应物离心分离,无水甲醇洗涤5次,所回收的CHT-Mn样品在烘箱80 ℃下干燥8 h,然后直接用于下一轮反应,如此反复使用4次。
2 结果与讨论
2.1 催化剂结构和织构表征结果
图1(a)为HT-0和HT-M前驱体的XRD谱图。由图1(a)可以看出,所有样品在2θ为11°、22°、35°、39°、46°、60°、62°处均呈现水滑石衍射峰,并没有检测到其他晶相,表明借助水滑石“记忆效应”,可将过渡金属离子引入水滑石层板,获得纯度较高的类水滑石前驱体。文献[10-12]报道,晶格参数a(=2d(110))和c(=3d(003))可反映类水滑石的六方晶系结构。表1为HT-0和HT-M前驱体材料的织构和化学性质。从表1可知,所合成的HT-M晶格参数符合文献报道范围,这说明HT-M具有规整的类水滑石结构[10-12]。

表1 HT-0和HT-M前驱体材料的织构和化学性质Table 1 The textural and chemical properties of HT-0 and HT-M precursors
图1(b)为CHT-0和CHT-M催化剂样品的XRD谱图。由图1(b)可以看出,经450 ℃焙烧后,样品的类水滑石特征峰均已消失,说明HT-0和HT-M材料热分解彻底,获得了CHT-0和CHT-M复合氧化物。同时,所有热分解产物均只出现了属于MgO(JCPDS 45-946)的衍射峰,并没有检测到Al2O3或M2Ox特征峰。通过计算2θ=43°左右的MgO衍射峰晶格参数可知,CHT-M的晶格参数均小于纯MgO(0.421 nm)[13],说明Al3+和Mx+均进入了MgO晶格,这有利于反应过程中催化剂结构稳定性的提高。

图1 HT-0、HT-M、CHT-0和CHT-M样品的XRD谱图Fig.1 XRD patterns of HT-0,HT-M,CHT-0 and CHT-M samples(a)HT-0 and HT-M;(b)CHT-0 and CHT-M
图2为CHT-0和CHT-M催化剂样品的N2吸附-脱附等温线和孔分布曲线。表2为CHT-0和CHT-M催化剂样品的织构和化学性质。由图2和表2可以看出,所有催化材料均有典型的Ⅳ型吸附-脱附曲线,并有明显的滞后环,且滞后环狭窄;同时,样品孔径均分布在5~40 nm之间,说明催化剂具有规整介孔结构[7-9]。更为重要的是,在焙烧过程中,类水滑石前驱体的分解产生了大量CO2和水蒸气。因此,与对应的前驱体相比,CHT-0和CHT-M具有较高孔体积和比表面积(见表1和表2)。

表2 CHT-0和CHT-M催化剂样品的结构、织构和碱性质Table 2 The structure,textural and basic properties of CHT-0 and CHT-M catalyst samples

图2 CHT-0和CHT-M催化剂样品的N2吸附-脱附等温线和孔分布曲线Fig.2 N2 adsorption-desorption isotherms and pore contribution of CHT-0 and CHT-M catalyst samples(a)N2 adsorption-desorption isotherms;(b)Pore contribution
HT-0和HT-M的SEM照片如图3所示。由图3可以看出,经过焙烧-重构过程,与呈现六边形形貌的HT-0相比,HT-M类水滑石前驱体形貌发生了明显的变化。Mn2+、Co2+、Cu2+、Zn2+等二价金属离子所改性的前驱体具有“花瓣”状形貌,并伴有碎片产生。与此不同的是,HT-Fe和HT-Cr形貌更为无序和扭曲。文献[13]报道,与二价金属阳离子相比,三价金属阳离子的溶解-再沉淀过程更为强烈。因而,在材料重构过程中,类水滑石形貌更为无序,团聚严重。

图3 HT-0和HT-M前驱体样品的SEM照片Fig.3 SEM images of HT-0 and HT-M precursors samples(a)HT-0;(b)HT-Cr;(c)HT-Mn;(d)HT-Fe;(e)HT-Co;(f)HT-Cu
2.2 催化剂表面碱性质和催化性能
众所周知,可借助CO2-TPD实验所测定的CO2脱附量和CO2最高脱附温度确定催化剂表面碱量和碱强度。脱附量越大、最高脱附温度越高,催化剂的碱量越大、碱强度越高[10-12]。CHT-0和CHT-M催化剂样品的CO2-TPD曲线如图4所示。可以看出,所有曲线都包含3个CO2脱附峰,分别是催化剂OH-基团产生的弱碱性位(<200 ℃),与金属-氧离子对(Mg-O、Al-O和M-O)有关的中强碱性位(200~400 ℃),以及由不饱和O2-离子产生的强碱性位(>400 ℃)[12]。过渡金属的加入,可调变CHT-0的表面碱性,尤其是催化剂强碱性位都有不同程度的右移,说明CHT-M具有较高的碱强度。以CO2最高脱附温度为序,催化剂碱强度从高到低的顺序为CHT-Co(490 ℃)、CHT-Mn(480 ℃)、CHT-Cu(460 ℃)、CHT-Cr(420 ℃)、CHT-Fe(410 ℃)、CHT-0(400 ℃)。由表2中CHT-0和CHT-M的表面碱量可知,添加过渡金属后,催化剂碱性位数量的变化与所掺杂金属有关。三价Cr3+和Fe3+离子的掺杂,对催化剂碱性位数量带来了不利影响;而二价离子的改性,可大幅提高Mg-Al复合氧化物的碱密度。尤其是CHT-Mn具有最高的碱性位数量(261 μmol/g),约为CHT-0样品的1.5倍。需要指出的是,在本研究中,催化剂表面碱量从高到低的顺序为CHT-Mn、CHT-Co、CHT-Cu、CHT-0、CHT-Fe、CHT-Cr。

图4 CHT-0和CHT-M催化剂样品的CO2-TPD曲线Fig.4 CO2-TPD profiles of CHT-0 and CHT-M catalyst samples
前期工作中,笔者研究了尿素和苯酚的反应网络(见图5)[10]。首先,尿素和苯酚作用得到目标产物水杨酰胺(SA),还有副产物4-羟基苯甲酰胺(HA)的生成(总反应A)。同时,水杨酰胺能进一步与苯酚反应生成呫吨酮(XA)(反应路径B)。此外,水杨酰胺会发生自缩合反应,脱水三聚成2,4,6-三(2-羟苯基)-1,3,5-均三嗪(HST)(反应路径C)。
表3为CHT-0和CHT-M催化合成水杨酰胺的反应性能。由表3可以看出:在无催化剂时,只有少量目标产物水杨酰胺生成,说明尿素与苯酚反应难以发生。以CHT-0为催化剂时,水杨酰胺收率为29.8%。对于CHT-M催化材料而言,催化效果取决于所掺杂过渡金属种类。CHT-Cr和CHT-Fe为催化剂时,水杨酰胺收率分别为18.7%和24.3%,低于CHT-0的催化性能。与CHT-Cr和CHT-Fe催化剂不同,CHT-Co和CHT-Cu催化剂的水杨酰胺收率可分别达到37.4%和34.9%,优于CHT-0的催化效果。而CHT-Mn复合氧化物,在相同反应条件下,水杨酰胺收率可达到41.2%。考虑到过渡金属改性可调变Mg-Al复合氧化物的表面碱性质,CHT-M催化性能可能与其碱强度和碱量有关。从上述讨论可知,与CHT-Co相比,CHT-Mn的碱强度较低,然而后者具有更优的催化性能。更为重要的是,所有催化剂的水杨酰胺收率大小顺序皆与其表面碱性位数量顺序相一致。因而,在制备水杨酰胺的反应过程中,催化剂活性可能取决于催化剂表面碱性位数量。为了验证上述猜想,笔者研究了反应起始2 h内的水杨酰胺收率和催化剂碱性位数量的关系,如图6所示。由图6可知,催化剂碱性位数量的增加有利于获得较高的水杨酰胺收率,说明催化剂催化能力的大小取决于其表面碱性位数量的高低。

(1)4-Hydroxybenzamide;(2)Salicylamide;(3)Bis(2-hydroxyphenyl)-methanone;(4)Xanthaone;(5)2,4,6-tris (2-hydroxyphenyl)-1,3,5-s-triazine图5 苯酚与尿素合成水杨酰胺可能的反应网络Fig.5 Possible reaction scheme of urea and phenol for the synthesis of salicylamide(a)The overall reaction of urea and phenol;(b)Xanthaone formation by reaction of salicylamide and phenol via removal of H2O and NH3;(c)Production of 2,4,6-tris(2-hydroxyphenyl)-1,3,5-s-triazine from dehydration of salicylamide
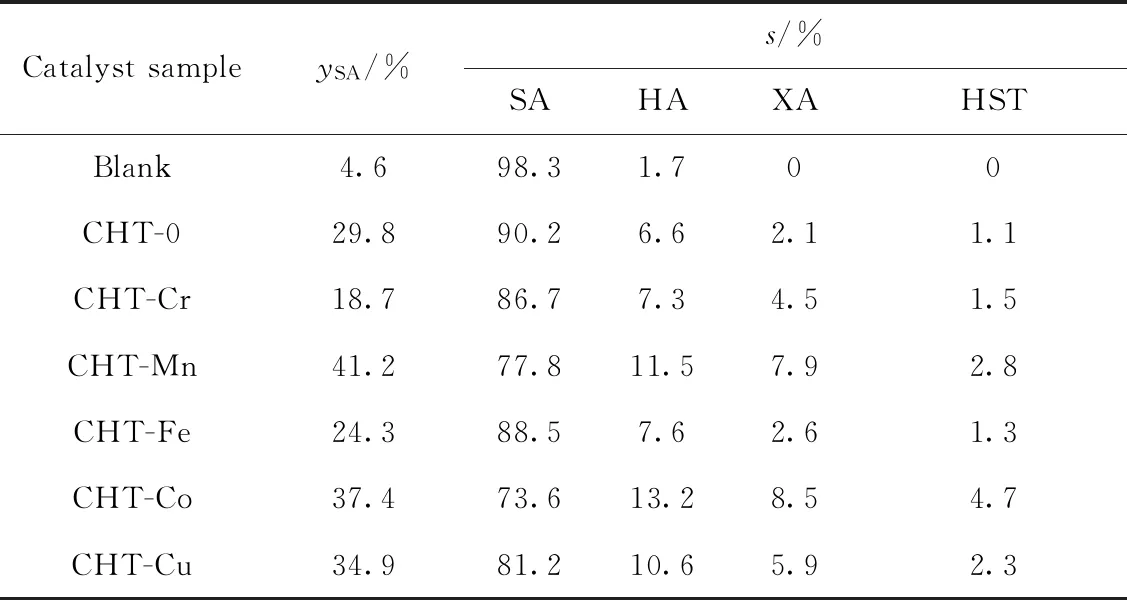
表3 CHT-0和CHT-M催化剂催化合成水杨酰胺的反应性能Table 3 Catalytic performance of CHT-0 and CHT-M catalysts for the synthesis of salicylamide
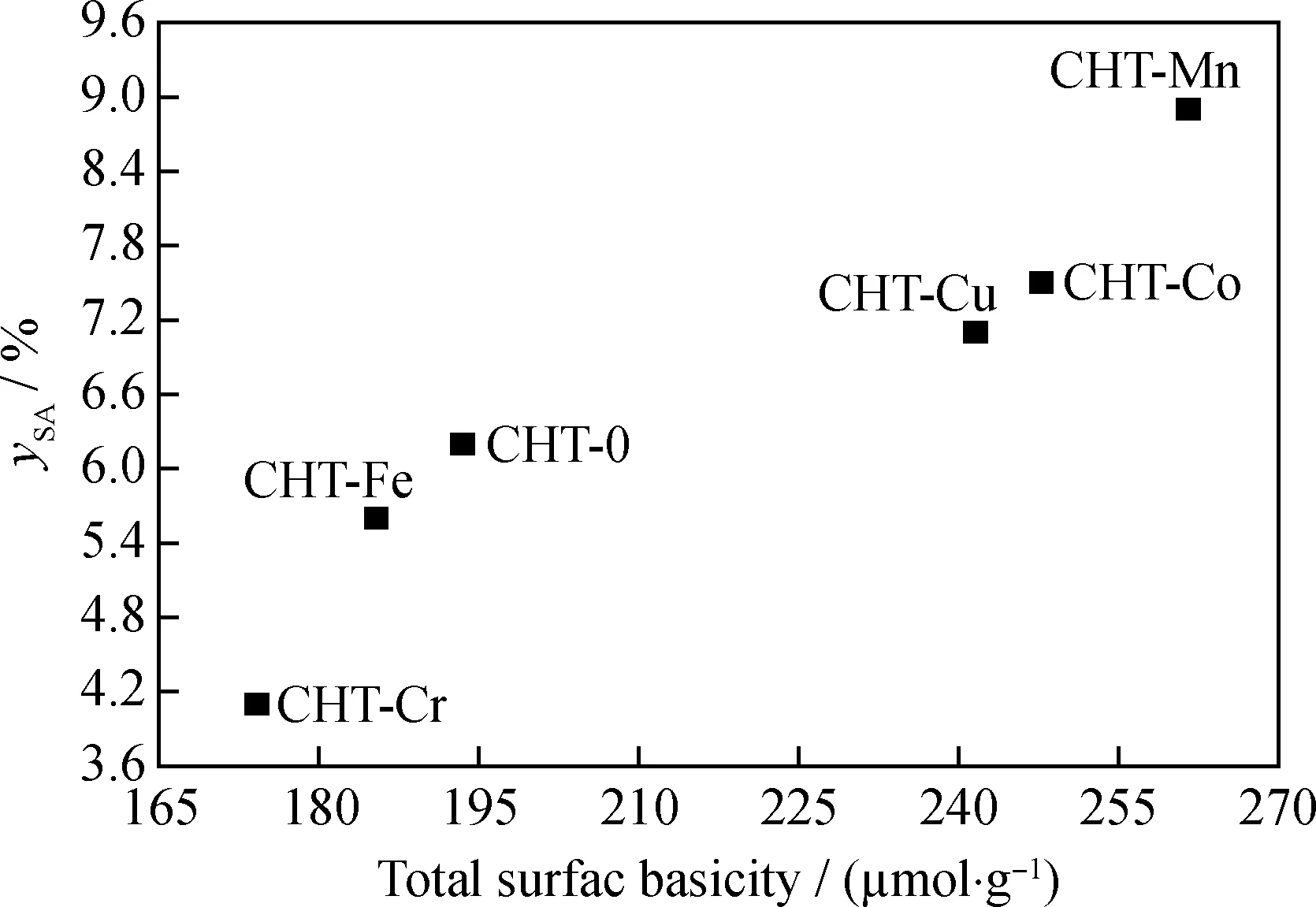
图6 CHT-0和CHT-M催化剂催化尿素和苯酚合成反应的水杨酰胺收率(ySA)与催化剂表面碱度之间的关系Fig.6 The relationship between the salicylamide yield (ySA)and the surface basicity for synthesis of salicylamide from urea and phenol over CHT-0 and CHT-M catalystsReaction conditions:T=210 ℃;t=2 h;m(Catalyst)=0.3 g;m(Urea)=3.0 g;m(Phenol)=23.5 g
然而,与单纯的Mg-Al复合氧化物(CHT-0)相比,过渡金属改性的CHT-M样品催化活性虽得到了提高,但是,它们的水杨酰胺选择性均低于CHT-0。经系统考察,发现水杨酰胺选择性随催化剂强碱性位碱强度的提升而降低(见图7)。文献[14]中报道,催化剂碱强度的提升有利于脱水反应的进行。这可能是由于催化剂碱性位可活化羟基氢原子(反应路径B)或氨基氢原子(反应路径C),增强了羟基氧原子或氨基氮原子的电负性,然后,氧原子或氮原子分别亲核进攻与其相邻的碳正原子,再经分子内电子重排,在形成新的C—O键或C=N键的同时,释放出H2O分子。本研究中,以水杨酰胺为原料的反应路径B和C都涉及有机物脱水反应分别生成副产物呫吨酮和2,4,6-三(2-羟苯基)-1,3,5-均三嗪(HST)。因此,具有强碱性位的催化剂会加剧副反应的发生,从而导致水杨酰胺选择性的降低。

图7 CHT-0和CHT-M催化剂催化尿素和苯酚合成反应的水杨酰胺选择性(sSA)与催化剂强碱性位脱附温度(碱强度)之间的关系Fig.7 The relationship between the salicylamide selectivity (sSA)and the desorption temperature of strong basic sites (basic strength)for synthesis of salicylamide from urea and phenol over CHT-0 and CHT-M catalystsReaction conditions:T=210 ℃;t=12 h;m(Catalyst)=0.3 g;m(Urea)=3.0 g;m(Phenol)=23.5 g
2.3 CHT-M催化水杨酰胺合成的可能机理
依据上述催化剂表面碱性表征和催化测试结果,结合文献报道,笔者提出了CHT-M催化尿素和苯酚合成水杨酰胺可能的催化机理,如图8所示。首先,尿素在碱性位作用下分解为氨气和异氰酸;同时,苯酚分子的酚羟基被催化剂碱性位吸附活化,在释放出氢质子的同时,转化为亲核的苯氧基负离子。然后,带负电的苯氧负离子亲核进攻异氰酸的碳原子;随之,苯环上与C—O相邻的C—H键断裂,氢原子转移到氮原子上,形成C—N键。最后,电子重排获得目标产物水杨酰胺。因此,催化剂碱性位数量越多,越有利于苯酚的活化,得到更多的酚羟基离子,使苯酚与尿素热分解产物异氰酸反应的活化能明显降低,有利于水杨酰胺的合成。因此,催化剂活性大小取决于其表面碱性位数目。CHT-Mn 的表面碱密度最大,该催化剂具有最强的催化能力。

图8 CHT-M催化尿素和苯酚合成水杨酰胺的可能机理Fig.8 Possible reaction mechanism towards the production of salicylamide from urea and phenol over CHT-M catalysts
2.4 反应条件对水杨酰胺合成的影响
以CHT-Mn为催化剂,考察了反应温度、反应时间和催化剂用量对水杨酰胺合成的影响,以期获得最佳反应条件。
2.4.1 反应时间的影响
反应时间对CHT-Mn催化剂催化合成水杨酰胺反应的影响如图9(a)所示。由图9(a)可以看出,在反应开始12 h内,水杨酰胺收率持续增加。然而,当反应时间超过12 h时,水杨酰胺收率出现了下降。因此,合成水杨酰胺的最适宜反应时间为12 h。
2.4.2 反应温度的影响
在反应时间12 h、催化剂用量0.3 g条件下,考察了反应温度对CHT-Mn催化剂催化合成水杨酰胺反应的影响(见图9(b))。尿素与苯酚反应是吸热反应,因而,随着反应温度升高,水杨酰胺收率持续增加;当反应温度为210 ℃时,水杨酰胺收率最高。进一步提高温度,使水杨酰胺自缩聚加剧和呫吨酮的生成,导致水杨酰胺收率的快速降低[3]。因此,水杨酰胺合成的最佳反应温度为210 ℃。
2.4.3 催化剂用量的影响
催化剂用量对CHT-Mn催化剂催化合成水杨酰胺反应的影响如图9(c)所示。由图9(c)可以看出,催化剂用量越大,所能提高的活性位数量越多,所以,随着CHT-Mn用量的增加,水杨酰胺收率随之提高。然而,当催化剂用量超过0.3 g时,水杨酰胺收率出现明显下降。这是由于,随着催化剂活性位的增加,副反应也剧烈发生,其反应速率超过了水杨酰胺生成的反应速率。因此,催化剂的最佳用量为0.3 g。

图9 反应时间(t)、反应温度(T)和催化剂用量(m(Catalyst))对CHT-Mn催化合成水杨酰胺反应的影响Fig.9 Effect of reaction time (t),reaction temperature (T) and catalyst mass (m(Catalyst))on the synthesis of salicylamide over CHT-Mn catalyst(a)ySA vs.t;T=210 ℃,m(Catalyst)=0.3 g,m(Urea)=3.0 g,m(Phenol)=23.5 g;(b)ySA vs.T;t=12 h,m(Catalyst)=0.3 g,m(Urea)=3.0 g,m(Phenol)=23.5 g;(c)ySA vs.m(Catalyst)T=210 ℃,t=12 h,m(Urea)=3.0 g,m(Phenol)=23.5 g
2.5 CHT-Mn的重复使用性
对多相催化剂而言,除催化剂活性外,其重复使用性也至关重要,因而,进行了CHT-Mn催化剂的再生性能实验。表4为CHT-Mn催化剂催化尿素和苯酚合成水杨酰胺的重复使用性数据。从表4可以看出,CHT-Mn经过4次再生重复使用后,催化活性变化不大,与新鲜催化剂相近。这说明该催化剂具有较好的可重复使用性,是一种具有工业前景的多相固体催化剂。

表4 CHT-Mn催化剂催化尿素和苯酚合成水杨酰胺的重复使用性Table 4 The reusability of CHT-Mn catalyst for the synthesis of salicylamide from urea and phenol
3 结 论
(1)借助水滑石“记忆效应”,成功地将过渡金属离子引入类水滑石层板中;再将它们煅烧,获得了一系列过渡金属改性Mg-Al复合氧化物介孔催化剂。
过渡金属离子种类对催化剂物化性质有较大影响,进而影响它们的碱强度和表面碱量。
(2)与催化剂碱强度相比,催化剂表面碱量对尿素和苯酚合成水杨酰胺反应影响较大。这是因为,催化剂表面碱量越大,越有利于苯酚的活化和水杨酰胺的生成。在反应温度210 ℃、反应时间12 h、CHT-Mn用量为0.3 g的适宜条件下,水杨酰胺收率可达到41.2%。
(3)CHT-Mn催化剂再生性能实验结果表明,该催化剂在重复使用4次后,催化活性基本保持不变,是一种廉价、易于制备的多相固体催化剂。
猜你喜欢
化学与生物工程(2022年9期)2022-09-30
环境工程技术学报(2022年3期)2022-06-05
能源化工(2021年6期)2021-12-30
合成技术及应用(2021年1期)2021-01-07
当代水产(2020年3期)2020-06-15
上海电力大学学报(2020年1期)2020-03-16
读书文摘(下半月)(2020年9期)2020-03-10
世界农药(2019年3期)2019-09-10
世界农药(2019年3期)2019-09-10
科学与财富(2019年3期)2019-02-28
