
多重PCR检测唾液标本中的幽门螺杆菌16S rRNA及cagA基因
2022-01-12 03:33:00王鑫莹孙丽媛杨志平宋顺佳田婉佳赵云冬
南方医科大学学报 2021年12期
王鑫莹,孙丽媛,杨志平,宋顺佳,李 南,刘 悦,田婉佳,赵云冬
北华大学1医学技术学院,2附属医院消化内科,吉林 吉林 132013;3吉林大学白求恩第一医院生殖中心-产前诊断中心,吉林 长春130021
幽门螺杆菌(Hp)与慢性胃炎、胃溃疡、十二指肠溃疡、胃粘膜相关组织性淋巴瘤及胃癌等疾病的发生密切相关[1-5]。1994年,世界卫生组织(WHO)已经将Hp列为第I类致癌因子[6]。Hp曾被认为是唯一能够在胃内生存的细菌[7],但随后有学者从胃液中培养了厚壁菌门、变形菌门和拟杆菌门等,上述微生物的发现揭示了上消化道可能存在一个复杂的细菌菌群,同时经研究发现Hp的存在会减少上消化道内物种的丰富性,并可能进一步影响细菌群落结构[8]。
目前检测Hp感染的方法中,侵入性的方法需要临床医生通过胃镜钳取活检组织,操作复杂,且患者多排斥;非侵入性的方法包括13C-尿素呼气试验、粪便Hp抗原检测、血液Hp抗体检测等,这些方法也均有各自的局限性。1989年Krajden等[9]首次报道了口腔中Hp的存在,并从Hp相关胃疾病患者的牙菌斑标本中成功分离培养出Hp。学者们推测口腔Hp可能是Hp根除率降低的原因之一,从胃中彻底根除Hp的困难可能与其在口腔中的存在有关[10]。刘经纬等[11]人通过PCR技术同时检测胃窦炎患者口腔及胃黏膜中的Hp,发现胃粘膜Hp阳性与阴性患者之间牙菌斑及唾液Hp的检出率存在显著差异,表明口腔内Hp与胃内Hp间存着一定的病因学联系。大量研究证明口腔内Hp感染与胃内Hp感染具有高度的相关性[12-14],且学者们发现口腔唾液标本、牙菌斑标本中Hp及胃活检标本中Hp具有高度的同源性[15-18],甚至基因型是相同的[19]。在一定的条件下,胃内的Hp会逆行进入口腔,口腔中Hp会随唾液进入胃内,Hp在胃及口腔中寄居,可能是胃疾病治疗后反复复发的主要原因[20-22],通过检测口腔中的Hp,可以对胃内Hp感染的诊断提供科学依据。
早期常采用分离培养的方法检测口腔中Hp,由于Hp培养条件苛刻、培养周期长等原因,其检测阳性率低。无论是感染前的筛查还是感染后的复查均可采用非侵入性的检测方法。近年来,基于聚合酶链反应(PCR)技术检测口腔内Hp的方法得到了广泛的研究,无论细菌的存活力如何都可以通过检测标本中Hp-DNA的基因靶点来检测Hp的存在,其具有较高的特异性及敏感性。细菌的分类和系统发育基于保守基因的序列,尤其是编码核糖体核糖核酸(rRNA)的16S rRNA基因。杨学文等[23]通过PCR方法扩增Hp的16S rRNA基因,对Hp临床分离菌株、胃活检组织以及其他非胃组织进行Hp检测,结果表明PCR方法扩增16S rRNA基因有较高的特异性。cagA基因为Hp公认的毒力标志基因,为关键的致病因子,其在Hp感染、致病以及致胃癌发生过程中发挥着重要的作用[24,25]。cagA阳性的Hp菌株与更高级别的胃炎有关,并且比cagA阴性菌株的毒力更强[26],且有研究表明,CagA是被认定的人类胃癌相关细菌肿瘤蛋白[27]。我国的Hp感染率很高,但是对于感染Hp的患者是否都需要治疗存在争议,由于患者不恰当的接受治疗会导致耐药性的增加,检测cagA基因可了解口腔中Hp是否携带毒力基因,可作为Hp应用抗生素清除的依据。本研究采用PCR技术检测胃肠道疾病患者口腔标本中Hp的16S rRNA基因及cagA毒力基因,预期判断患者Hp感染及患病情况。
1 资料和方法
1.1 材料
1.1.1 一般资料 2019年7月~2020年8月因上消化道症状来北华大学附属医院门诊或住院的患者,经胃镜检查或病理检测确诊为Hp感染的胃、十二指肠疾病患者共156例,其中男性64例,女性92例,年龄18~87(46±1)岁,所有纳入实验的患者均对本研究知情并签订知情同意书。并经北华大学附属医院医学伦理委员会审查[2019年第(8)号]。
1.1.2 菌株 Hp标准菌株:ATCC26695、大肠埃希菌、金黄色葡萄球菌、直弯曲菌、唾液链球菌、乳酸杆菌均为本实验室保存菌种。
1.1.3 主要试剂与培养基 Columbia Agar Base(OXOID);脱纤维绵羊血(Solarbio);厌氧培养袋、微需氧产气包均购自青岛高科技工业园海博生物技术有限公司;TIANamp Bacteria DNA Kit、TIANamp Swab DNA Kit、质粒小提试剂盒、pGM-T载体连接试剂盒、琼脂糖凝胶DNA回收试剂盒、TIANSeq高保真PCR反应预混液、TaqPCR Mastermix、D2000均购自天根生化科技(北京)有限公司;Tris-base、硼酸、0.5M EDTA(pH8.0)均购自生工生物工程(上海)股份有限公司;核酸染料为StarGreen DNA Dye 10,000×购自上海开放生物科技有限公司。
1.2 方法
1.2.1 菌株复苏及基因组的提取 将实验室冻存菌液滴加到配制的哥伦比亚血平板上培养3~5 d,取复苏成功的Hp,用生理盐水将细菌悬浮成液,采用试剂盒(TIANamp Bacteria DNA Kit)提取Hp 基因组DNA。同时提取大肠埃希菌、金黄色葡萄球菌、直弯曲菌、唾液链球菌、乳酸杆菌基因组用于验证引物特异性,为避免交叉污染,提取时在不同的生物安全柜进行。
1.2.2 引物设计与合成 根据RefSeq及GenBank数据库中Hp16S rRNA及cagA基因序列,用DNAMAN软件设计特异性引物。经Blast软件对比分析后,筛选最优的两套引物。引物序列为HP-16s:F-ACGC ATAGGTCATGTGCCTC、R-GTGTCCGTTCACCCT CTCAG,扩增片段长度为182 bp;HP-cagA :FCTGGTGGGGATTGGCTTGAT、R-ACCAACGGTAG CGTTCCAAT,扩增片段长度为401 bp。引物由生工生物工程(上海)股份有限公司合成。
1.2.3 引物特异性试验 应用Hp及大肠埃希菌、金黄色葡萄球菌、直弯曲菌、唾液链球菌、乳酸杆菌的基因组作为模板,进行PCR反应。
1.2.4 引物灵敏度试验 稀释Hp DNA至100 ng/μL左右,以102、10、1、10-1、10-2、10-3、10-4ng·μL-17个梯度浓度的基因组为模板进行PCR反应验证引物Hp-16s及Hp-cagA的灵敏性。
1.2.5 多重PCR建立25μL反应体系,其中引物Hp-16s-F及引物Hp-16s-R各0.5 μL、引物Hp-cagA-F及引物HpcagA-R各0.75 μL、2×Taq Master Mix 12.5 μL、模板1 μL、ddH2O 9 μL。反应条件:94 ℃预变性5 min,94 ℃30 s、59 ℃30 s、72 ℃30 s,30个循环,72 ℃延伸10 min。
1.2.6 多重PCR灵敏性试验 以102、10、1、10-1、10-2、10-3、10-4ng·μL-17个梯度浓度的基因组为模板进行多重PCR反应。
1.2.7 重组阳性质粒的构建 ①PCR扩增目的基因分别以引物Hp-16s及Hp-cagA为上下游引物,以Hp标准菌株为扩增模板。进行定性PCR扩增,反应体系为引物Hp-16s-F及引物Hp-16s-R各0.5 μL或引物Hp-cagA-F及引物Hp-cagA-R各0.5μL、2×Taq Master Mix 12.5μL、模板1 μL、ddH2O 10.5 μL,反应条件见同1.2.5;②应用天根普通琼脂糖凝胶DNA回收试剂盒纯化PCR产物;③采用pGM-T 载体连接试剂盒将回收的片段与载体pGM-T进行连接;④将连接产物转化到感受态细胞中,经蓝白斑筛选与鉴定后,应用质粒小提试剂盒提取重组质粒DNA(分别命名为pGMT-16s及pGMT-cagA),最后由生工生物工程(上海)股份有限公司进行测序。
1.3 唾液标本的收集及提取其基因组DNA
1.3.1 唾液标本收集 患者清晨空腹(未刷牙及漱口),操作人员戴着无菌手套手持无菌痰盒,嘱患者自然流出唾液,禁止咳痰,及时盖紧做好标记并用封口膜封住管口,以防止交叉污染。
1.3.2 提取唾液标本DNA 取500 μL唾液到Ep管中,并用生理盐水悬浮,10 000 r/min离心1 min,尽量吸净上清,收集菌体。试剂盒优化:加入400 μL Buffer GA,20 μL Proteinase K 旋涡混匀后56 ℃放置60 min,其间每15 min 涡旋混匀数次。之后按照试剂盒(TIANamp Swab DNA Kit)说明书操作,最后用缓冲液TB 溶解后-20 ℃储存备用。
1.4 多重PCR检测唾液标本Hp
25 μL 反应体系,其中引物Hp-16s-F 及引物Hp-16s-R各0.5 μL、引物Hp-cagA-F及引物Hp-cagA-R各0.75 μL、2×Taq Master Mix 12.5 μL、模板5 μL、ddH2O 5 μL,反应条件同1.2.5,同时设阴性对照和阳性对照,阳性对照模板为构建的重组质粒,反应体系为引物Hp-16s-F及引物Hp-16s-R各0.5 μL、引物Hp-cagA-F及引物Hp-cagA-R各0.75 μL、2×Taq Master Mix 12.5 μL、pGMT-16s 0.5 μL、pGMT-cagA 0.5 μL、ddH2O 9 μL。
1.5 琼脂糖凝胶电泳
取10 μL PCR 产物于1.2%琼脂糖凝胶电泳,StarGreen染色,以D2000 DNA marker作为DNA分子量标准,用凝胶电泳成像系统进行分析。
2 结果
2.1 引物特异性试验
Hp在相应位置产生特异的DNA条带,引物Hp-16s扩增出182 bp片段大小条带(图1),引物Hp-cagA扩增出401 bp片段大小条带(图2)。大肠埃希菌、金黄色葡萄球菌、直弯曲菌、唾液链球菌、乳酸杆菌均未扩增出条带,表明引物Hp-16s及Hp-cagA特异性良好。
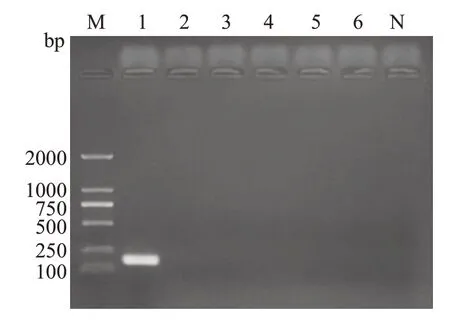
图1 引物Hp-16s特异性试验Fig.1 Primer Hp-16s specificity test.M:D2000 DNA Marker;N:Blank;Lane 1:Hp standard strain;Lane 2: Escherichia coli;Lane 3: Staphylococcus aureus;Lane 4: Campylobacter rectus;Lane 5: Streptococcus salivarius;Lane 6:Lactobacillus.
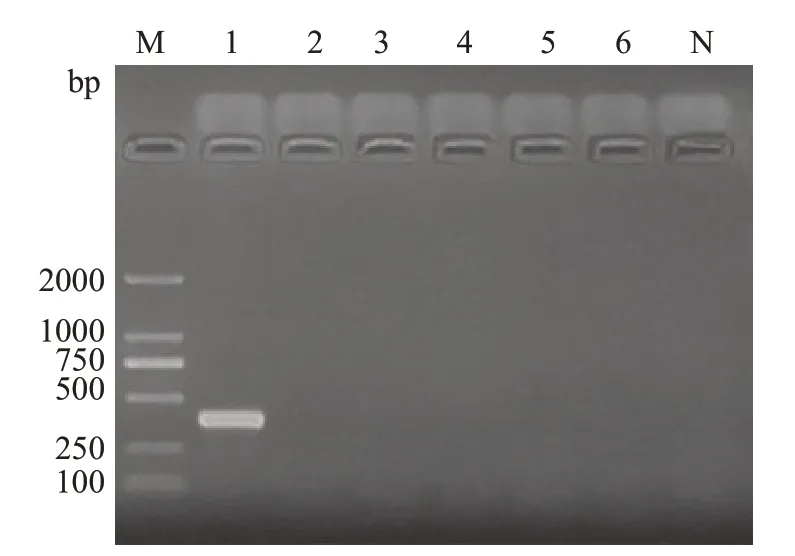
图2 引物Hp-cagA特异性试验Fig.2 Primer Hp-cagA specificity test.M:D2000 DNA Marker;N Blank;Lane 1:Hp standard strain;Lane 2: Escherichia coli;Lane 3: Staphylococcus aureus;Lane 4: Campylobacter rectus;Lane 5: Streptococcus salivarius;Lane 6:Lactobacillus.
2.2 引物灵敏性试验
当模板浓度为10-3ng·μL-1(计算得拷贝数5.46×102copies·μL-1)时,引物Hp-16s仍能扩增出182 bp片段大小条带(图3),同时当模板浓度为10-3ng·μL-1时,引物Hp-cagA仍能扩增出401 bp片段大小条带(图4),该方法最低能检出102copies·μL-1Hp16S rRNA或cagA基因。
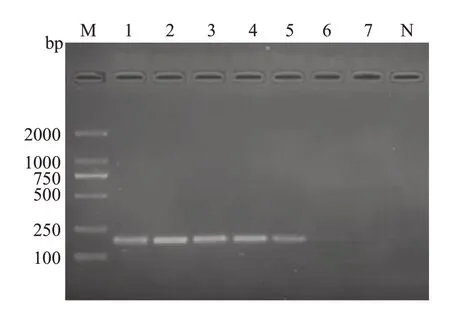
图3 引物Hp-16s灵敏性试验Fig.3 Primer Hp-16s sensitivity test.M:D2000 DNA Marker;N Blank;Lane 1:100 ng•μL-1;Lane 2:10 ng•μL-1;Lane 3:1 ng•μL-1;Lane 4:1×10-1 ng•μL-1;Lane 5:1×10-2 ng•μL-1;Lane 6:1×10-3 ng•μL-1;Lane 7:1×10-4 ng•μL-1.

图4 引物Hp-CagA灵敏性试验Fig.4 Primer Hp-CagA sensitivity test.M:D2000 DNA Marker;N:Blank;Lane 1:100 ng•μL-1;Lane 2:10 ng•μL-1;Lane 3:1 ng•μL-1;Lane 4:1×10-1 ng•μL-1;Lane 5:1×10-2 ng•μL-1;Lane 6:1×10-3 ng•μL-1;Lane 7:1×10-4 ng•μL-1.
2.3 多重PCR建立
只有带有cagA基因的Hp扩增出182 bp及401 bp片段两条条带,大肠埃希菌、金黄色葡萄球菌、直弯曲菌、唾液链球菌、乳酸杆菌均为产生条带,该方法能检测Hp16S rRNA及cagA基因。
2.4 多重PCR灵敏性试验
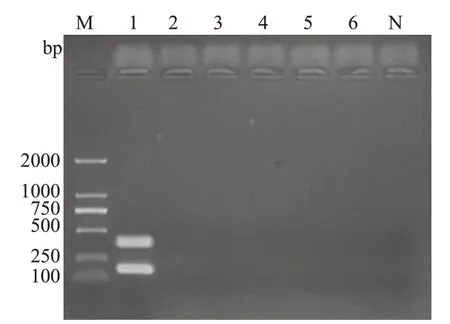
图5 多重PCR凝胶电泳图谱Fig.5 Multiplex PCR gel electrophoresis pattern.M:D2000 DNA Marker;N:Blank;Lane 1:Hp standard strain;Lane 2:Escherichia coli;Lane 3: Staphylococcus aureus;Lane 4: Campylobacter rectus;Lane 5:Streptococcus salivarius;Lane 6:Lactobacillus.

图6 多重PCR灵敏性试验Fig.6 Multiplex PCR sensitivity test.M:D2000 DNA Marker;N:Blank;Lane 1:100 ng•μL-1;Lane 2:10 ng•μL-1;Lane 3:1 ng•μL-1;Lane 4:1×10-1 ng•μL-1;Lane 5:1×10-2 ng•μL-1;Lane 6:1×10-3 ng•μL-1;Lane 7:1×10-4 ng•μL-1.
以102、10、1、10-1、10-2、10-3、10-4ng·μL-17个梯度浓度的基因组模板进行多重PCR 检测,当模板浓度为10-2ng·μL-1(计算得拷贝数5.46×103copies·μL-1)时,仍能检测出Hp 16S rRNA及cagA基因(图2),本研究方法最低检测线为103copies·μL-1。
2.5 克隆目的条带、测序比对结果
重组质粒经PCR反应、凝胶电泳后均在相应位置出现特异性条带,与预期目的条带位置一致(图7)。测序结果经Blast比对分析,pGMT-16s与GenBank中已登记的Hp16S rRNA基因相似性为100%,pGMT-cagA与GenBank中已登记的HpcagA基因序列的相似性为100%。
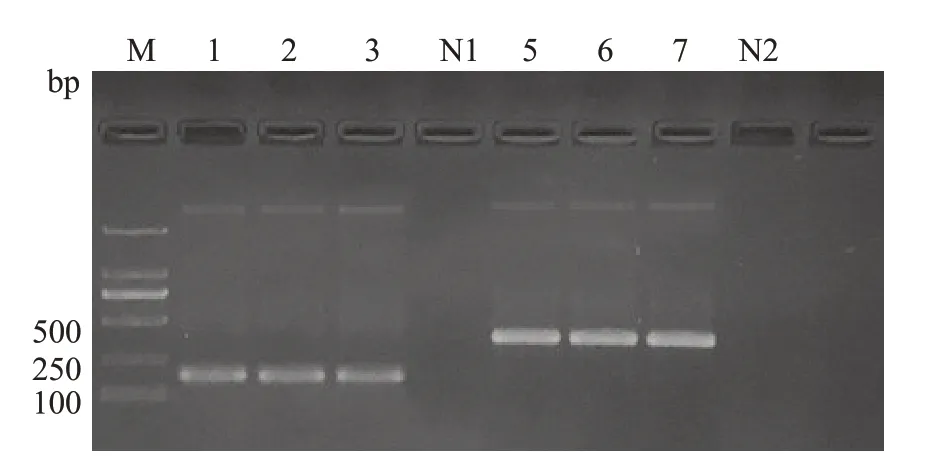
图7 重组质粒PCR电泳结果Fig.7 Recombinant plasmid PCR electrophoresis results.M:D2000 bp DNA Ladder;N1-N2:Blank;Lanes 1-3:pGMT-16s;Lanes 5-7:pGMT-cagA.
2.6 消化道疾病患者唾液标本多重PCR扩增检测
在156例消化道疾病患者中,有136例唾液标本通过多重PCR方法扩增出Hp16S rRNA基因,36例扩增出HpcagA基因。其中,有36例16S rRNA+/cagA+,100例16S rRNA+/cagA-,20例16S rRNA-/cagA-,部分琼脂糖凝胶电泳结果见图8。
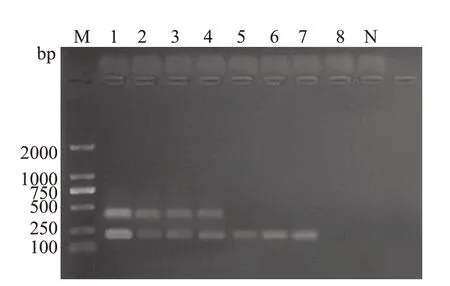
图8 多重PCR扩增临床唾液标本Hp16S rRNA及cagA基因凝胶电泳图谱Fig.8 Gel electrophoresis profiles of multiplex PCR amplification of Hp 16S rRNA gene and cagA gene in clinical saliva samples.M:D2000 DNA Marker;N:Blank;Lane 1:pGMT-Hp;Lane 2:Saliva specimen No.14;Lane 3:Saliva specimen No.60;Lane 4:Saliva specimen No.136;Lane 5:Saliva specimen No.22;Lane 6:Saliva specimen No.96;Lane 7:Saliva specimen No.120;Lane:8 Saliva specimen No.7.
3 讨论
目前我国人群Hp感染率约达50%[28]。Hp感染在许多胃部疾病的发病机理中起着重要作用。由于感染的存在与否决定了临床不同的治疗措施,所以对Hp感染的检测已成为胃和十二指肠疾病诊断过程中非常重要的部分。尽管人们对于Hp感染的重视程度在提高,而口腔Hp的存在很容易被人们忽视,因此建立一种快速、方便检测口腔Hp的方法尤为重要。
本文通过RefSeq及GenBank数据库获取Hp16S rRNA及cagA基因序列,利用DNAMAN 软件对比分析,设计特异性引物,对常见与Hp容易出现交叉反应的细菌进行引物特异性实验,最适退火温度为59 ℃时,只有Hp标准菌株在相应位置出现特异性条带,其他细菌均未产生条带。对模板浓度系列稀释后进行引物灵敏性试验,该方法最低可检测102copies·μL-1Hp16S rRNADNA或HpcagADNA,建立检测Hp的16S rRNA及cagA基因的多重PCR方法,扩增的目的片段分别为182 bp 和401 bp。建立的多重PCR 方法可同时检测Hp16S rRNA及cagA基因,该多重PCR体系中所用酶量与普通PCR基本相同,因此节省了试剂,减轻了患者负担。通过多重PCR灵敏性检测结果表明,当模板浓度为1×10-2ng·μL-1(计算得拷贝数5.46×103copies·μL-1)时,仍能在相应位置检测到条带。该方法最低检测线为103copies·μL-1,因此该方法特异性及灵敏性良好。
同时应用分子克隆技术构建重组质粒pGMT-16s及pGMT-cagA,经测序比对分析后,与GenBank中已登记的Hp 16S rRNA(GenBank:MN326691.1)及Hp cagA(GenBank:AB246743.1)同源性均为100%,与其他细菌无匹配,可作为Hp DNA标准阳性对照品。构建的重组质粒易于保存,稳定性良好。
通过建立的多重PCR方法检测Hp感染的消化道疾病患者的口腔唾液标本,口腔携带Hp16S rRNA基因的阳性率高达87.2%,cagA基因阳性率为23.1%。因此Hp的清除不仅应针对胃内,同样应重视对口腔中Hp的清除。通过检测口腔标本中的cagA基因的结果发现,不存在cagA基因单独阳性的结果,所有cagA基因阳性的标本,16S rRNA基因均为阳性,与Ansari等[29]的研究结果一致。本研究检测的cagA基因阳性患者中多数患有消化性溃疡疾病,且有2例胃癌患者。因此cagA基因可联合16S rRNA基因检测,实现对Hp感染患者的初步预测,可指导临床合理用药。在一些接受抗菌药物、铋剂等治疗的住院患者(52例)中,本方法同样检测到了Hp的存在,说明该方法不受上述药物的影响。因此采用分子生物学方法检测Hp可不必停药(碳呼气试验检测前需要停药1月以上),随着抗生素耐药的不断增加,进行Hp治疗的患者应确认是否根除。该方法对患者治疗后的复查有很大的帮助。
随着现代医疗技术的发展,无创的检测方法更受患者青睐,唾液诊断作为一种新兴的诊断技术现已成为研究的前沿[30]。检测唾液标本Hp作为一种非侵入性检测的方法,标本采集方便,获得容易,进行口腔Hp的检测为预防Hp相关疾病的发生发展起着积极、重要的作用。尽管唾液标本中Hp的含量较低,但应用本方法仍可准确的检测出Hp的存在并判断其是否携带毒力因子cagA基因,更重要的是检测结果不受近期是否服用过抗生素、质子泵抑制剂等药物的限制,因此在诊断Hp相关性胃部疾病有良好的研发前景。
猜你喜欢
青少年科技博览(中学版)(2023年1期)2023-03-17 00:44:34
世界科学技术-中医药现代化(2022年9期)2023-01-17 07:39:18
黄河·黄土·黄种人(华夏文明)(2021年6期)2021-09-28 02:14:08
现代仪器与医疗(2021年1期)2021-06-09 05:53:54
中国生殖健康(2020年8期)2021-01-18 03:05:20
中国生殖健康(2018年3期)2018-11-06 07:20:02
读者·校园版(2017年9期)2017-04-15 13:51:36
系统工程与电子技术(2016年2期)2016-04-16 05:16:53
中国光学(2015年1期)2015-06-06 18:30:20
海岸工程(2014年4期)2014-02-27 12:51:28
