
Pickering双乳液法制备分子印迹聚合物分离分析玉米中的硝磺草酮
2022-01-09 06:46葛俊康柏晓丽郭昕鹭祁烁霖陈立钢
分析测试学报 2021年12期
葛俊康,柏晓丽,郭昕鹭,祁烁霖,牛 娜,陈立钢
(东北林业大学 化学化工与资源利用学院,黑龙江 哈尔滨 150040)
硝磺草酮(Mesotrione)是一种可以抑制对羟苯基丙酮酸双氧化酶活性的选择性除草剂[1-2],过量施加会给农作物安全带来潜在危害,并通过食物链传递对人体健康产生影响[3],因此检测食品中残留硝磺草酮的含量尤为重要。硝磺草酮在高温条件下不稳定,常用液相色谱法进行检测,气相色谱法进行分析时须采用间接法[4],且在色谱分析前,必须经过操作复杂、过程繁琐的样品前处理过程,如固相萃取等[5]。
采用基质固相分散(MSPD)技术萃取分析玉米中的硝磺草酮,其提取、净化和过滤可一步完成,减少了溶剂消耗量,简化了分析步骤[6-7]。传统的MSPD 分散剂在分散复杂样品时,存在富集效果差、特异性低等问题[8]。分子印迹聚合物(Molecularly imprinted polymer,MIP)是一种对目标分子具有选择性结合能力的材料,采用MIP 作为MSPD 的分散剂,可以提高对化合物的亲和性,降低干扰物产生的影响[9]。
传统制备MIP 的方法主要为本体聚合,此方法操作过程繁琐,制备的MIP 形貌不规则[10]。与传统制备方法相比,Pickering 双乳液法制备的MIP 具有形貌规则,模板分子易洗脱等优点[11-12]。在材料制备中,可引入价廉易得[13]的木质素作为稳定粒子。木质素经过油酸改性处理后,可得到稳定的W/O Pickering 乳液粒子,亲水性木质素与疏水性木质素可以在界面形成无表面活性剂的稳定的W1/O/W2Pickering双乳液[14]。
本研究以硝磺草酮为模板分子,甲基丙烯酸甲酯为功能单体,木质素为稳定粒子,采用Pickering双乳液技术制备了硝磺草酮MIP,并通过动态吸附、静态吸附及选择性吸附,研究了其吸附行为。将MIP用作MSPD的分散剂对样品进行处理,并通过高效液相色谱测定了玉米样品中硝磺草酮的含量。本文的前处理方法快速,具有较高的选择性。
1 实验部分
1.1 仪器与试剂
LC-15C 高效液相色谱仪、XRD-600 X 射线衍射仪(日本岛津公司);Nicolet360 傅立叶变换红外光谱仪(美国尼高力仪器公司);JEM-6700 F 扫描电镜(捷欧路科贸有限公司);OCA 20 接触角测量仪(北京奥德利诺有限公司);KQ5200E 超声波清洗器(昆山舒美超声仪器有限公司);HSY-B 恒温振荡摇床(常州金坛精达仪器制造有限公司)。
木质素(98%,山东龙力生物科技股份有限公司);莠去津(98%,上海阿拉丁生化科技股份有限公司);硝磺草酮(97%)、磺草酮(98%)(上海源叶生物科技有限公司);色谱纯乙腈(成都思为科学仪器有限公司);甲基丙烯酸甲酯、偶氮二异丁酸二甲酯、二乙烯基苯、苯乙烯、油酸、冰醋酸、乙醇(天津市科密欧化学试剂有限公司),以上试剂均为分析纯;实验用水为去离子水;玉米样品购于哈尔滨当地超市。
1.2 实验方法
1.2.1 分子印迹聚合物的制备准确称取3.0 g木质素,加入30 mL去离子水中以形成木质素分散体,以浓氨水调至pH 11.0,待木质素完全溶解后,用1 mol/L 盐酸调至pH 3.0,过滤并干燥,得亲水性木质素。精密量取49 mL 苯乙烯溶液,加入2.0 g 亲水性木质素,超声处理3 min 后,再加入1 mL 油酸,并于50 ℃恒温下磁力搅拌12 h,获得疏水性木质素。取0.5 mL 甲基丙烯酸甲酯、1.5 mL 二乙烯基苯、40 mg 偶氮二异丁酸二甲酯和0.4 g 硝磺草酮溶于2.0 mL 疏水性木质素中,添加1 mL 0.1 mol/L NaCl 溶液和4.0 mL亲水性木质素分散液(称取1.0 g亲水性木质素,分散在100 mL去离子水中,形成亲水性木质素分散液),得W1/O/W2双乳液,于65 ℃下反应12 h,用乙醇和水洗涤未反应产物并干燥,再经乙酸-甲醇溶液(体积比15∶85)索氏提取除去产物中的模板分子,干燥得MIP,制备过程如图1所示。

图1 MIP的制备过程示意图Fig.1 Schematic illustration of the preparation process of MIP
非分子印迹聚合物(NIP)的制备不添加模板分子,其余步骤与上述相同。
1.2.2 吸附实验静态吸附实验:将20 mg MIP(或NIP)加入8 mL 不同质量浓度(5、10、20、50、80、100、120、150、200µg/mL)硝磺草酮标准溶液中并设置空白组,于25 ℃恒温水浴中振荡24 h;动态吸附实验:将20 mg MIP(或NIP)加入9 组8 mL 200µg/mL 的硝磺草酮标准溶液中并设置空白组,于25 ℃恒温水浴中振荡不同时间(1、2、5、10、20、30、45、60、90 min);选择性吸附实验:精确称取3份20 mg MIP(或NIP),分别加入8 mL 200µg/mL硝磺草酮标准溶液、莠去津标准溶液、烟嘧磺隆标准溶液中,于25 ℃恒温水浴中振荡24 h。吸附后溶液以7 000 r/min 离心5 min,取上清液过滤,进行HPLC检测。
1.2.3 玉米中硝磺草酮的基质固相分散萃取精确称取0.2 g玉米样品和0.3 g MIP加入玛瑙研钵中研磨10 min,再取已装有玻璃棉垫底的萃取柱(5 mL),将研磨好的样品放入萃取柱,样品上方放置适量玻璃棉并压实。以2 mL 20%甲醇水溶液淋洗,并用5 mL 5%乙酸乙腈溶液洗脱目标物,流速为1 mL/min,洗脱液用氮气吹干后于0.5 mL色谱流动相中溶解,待测。
1.2.4 色谱条件Hypersil ODS2色谱柱(4.6 mm×150 mm,5µm);流动相:乙腈-水(体积比85∶15);流速1.0 mL/min;进样体积20µL;检测波长233 nm。
2 结果与讨论
2.1 分子印迹聚合物的表征
MIP 的傅里叶红外光谱及扫描电镜图见图2。798 cm-1和882 cm-1处的吸收峰为C—H 苯环的弯曲振动;1 608 cm-1处的吸收峰由C—C 苯环的振动所产生,说明二乙烯基苯成功参与了印迹聚合反应。1 745 cm-1处的吸收峰代表C====O 拉伸振动,说明甲基丙烯酸甲酯成功参与反应。在波长3 137 cm-1和3 622 cm-1位置的强振动表明木质素含有苯酚和脂肪族羟基(图2A)。而MIP和NIP具有类似的红外光谱图,表明模板分子已成功洗脱。由扫描电镜图可以看出MIP 形貌规则,且颗粒尺寸较大(图2B),有助于基质固相分散过程,避免柱压过高。
对制备的亲水性木质素、疏水性木质素及MIP 的接触角进行测量(图2C),结果发现三者的接触角大小分别为71.5°、99.9°、149.3°,这是因为未改性的木质素亲水,有助于形成水包油乳液;而油酸吸附到木质素表面后,使得木质素具有疏水性;在聚合反应后,由于二乙烯基苯和甲基丙烯酸甲酯的加入,致使MIP的接触角最大,其疏水性表面有利于吸附低水溶性且疏水的硝磺草酮[15]。
Pickering双乳液法制备的MIP的XRD光谱图在2θ=20°处出现一个光滑的衍射峰,表明该MIP是无定形物质(图2D)。

图2 MIP的红外光谱图(A)和扫描电镜图(B),亲水性、疏水性木质素以及MIP的接触角(C)与MIP的X射线衍射谱图(D)Fig.2 Infrared spectrum(A)and scanning electron microscope(B)of MIP,contact angle of hydrophilic,modified hydrophobic lignin and MIP(C),and X-ray diffraction pattern of MIP(D)
2.2 吸附等温线的测定与Scatchard分析
由图3A 可见,无论是MIP 还是NIP,对硝磺草酮的吸附量均随其浓度递增,但在相同浓度下,MIP的吸附效果远高于NIP,表明MIP上所形成的分子印迹空穴使其特定识别和吸附模板分子的能力增强。采用Scatchard分析聚合物的吸附情况[16],公式如下:

图3 MIP和NIP的吸附等温线(A),MIP(B)和NIP(C)的Scatchard分析曲线Fig.3 Adsorption isotherms of MIP and NIP(A),Scatchard analysis curves of MIP(B)and NIP(C)

式中,Q为聚合物吸附硝磺草酮的容量大小(mg/g);C为平衡时硝磺草酮的质量浓度(µg/L);Qmax为聚合物吸附硝磺草酮的最大容量(mg/g);Kd为结合位点的解离常数(mg/L)。
以Q/C对Q作图得Scatchard 分析曲线,如图3B 所示,MIP 的Scatchard 分析曲线具有两种不同的斜率,表明MIP对硝磺草酮的吸附具有两种不同的结合位点,由两条拟合直线可求得截距和斜率分别为:Qmax1= 32.31 mg/g,Kd1= 116.28 mg/L;Qmax2= 89.99 mg/g,Kd2= 413.22 mg/L;则本实验制备的MIP 对硝磺草酮的吸附容量(Qmax1+Qmax2)为122.30 mg/g。此外,NIP 的Scatchard 分析曲线(图3C)具有良好的线性关系,表明NIP 对于硝磺草酮的吸附是均匀的,仅存在一个结合位点。由拟合直线的截距和斜率求得:Qmax=31.62 mg/g,Kd=270.27 mg/L。比较实验结果可知,MIP的最大表观结合量远高于NIP。
2.3 吸附动力学曲线的测定
从图4A 可知,MIP 和NIP 对硝磺草酮的吸附能力随时间延长而增强,而MIP 的硝磺草酮吸附量始终大于NIP。由于分子印迹孔穴较多,最初的40 min 内MIP 的吸附率很高,而60 min 后,大多数印迹孔穴已被填充,因此MIP 的吸附速率达到平衡和稳定。40~60 min 的快速平衡表明MIP 结合位点对硝磺草酮具有良好的亲和力。

图4 MIP和NIP的动力学吸附(A)、准一级动力学(B)和准二级动力学(C)Fig.4 Kinetic adsorption(A),quasi first order kinetics(B)and quasi second order kinetics(C)of MIP and NIP
分别采用准一级、准二级动力学反应对MIP和NIP进行拟合计算[17-18],拟合方程如下:

式中,Qt为不同时间的吸附量(mg/g);Qe为平衡时的吸附量(mg/g);k1、k2分别是准一级、准二级动力学的速率常数(min-1)。
通过ln(Qe-Qt)对t拟合计算,获得准一级动力学方程,如图4B所示,MIP和NIP的线性方程分别为y=-0.008 7x+3.435,r2=0.879 1;y=-0.014 9x+2.577 2,r2=0.781 3;以t/Qt对t拟合可获得如图4C所示的MIP、NIP 的准二级动力学方程:y=0.041 8x+0.912 7,r2=0.999 1;y=0.075 8x+2.158 9,r2=0.999 9。由准一级和准二级拟合曲线的r2分析比较可得,MIP和NIP吸附硝磺草酮的过程较切合准二级动力学模型。
2.4 吸附选择性的研究
本实验采用磺草酮为类似物,莠去津为参照物,探究MIP的选择性吸附性能,其分子结构如图5A所示。结合图5B 及表1数据可见,在吸附硝磺草酮的过程中,MIP 对硝磺草酮的吸附能力远大于NIP,表明制备的MIP对硝磺草酮具有更好的选择吸附性;MIP对磺草酮的吸附能力也明显高于NIP,这归因于磺草酮和硝磺草酮的结构相似。为了深入研究MIP的选择能力,通过公式(4)、(5)、(6)计算选择性参数;得其分配系数(δ)、选择因子(α)和相对选择因子(β)关系如下:
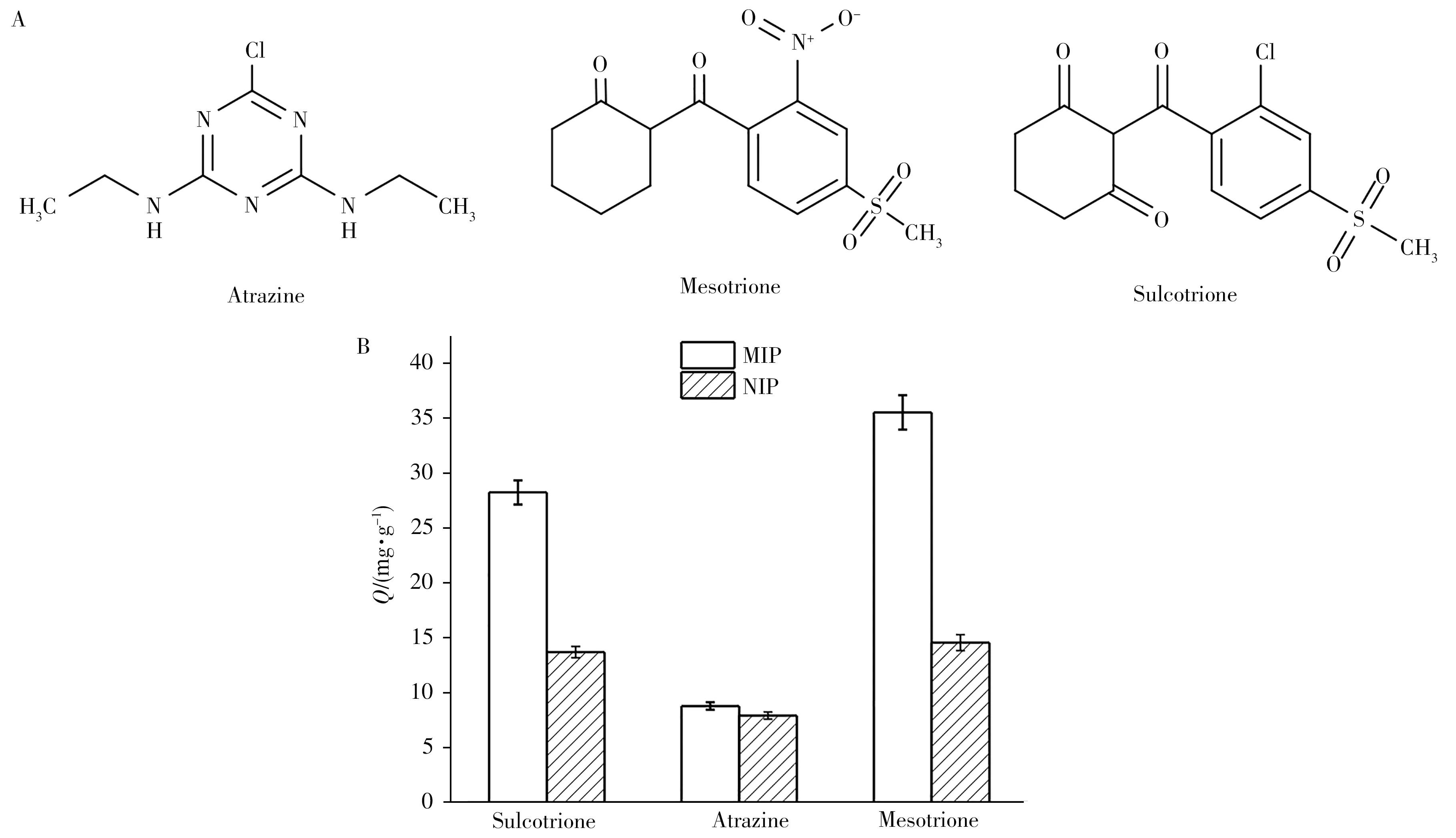
图5 莠去津、硝磺草酮、磺草酮的结构式(A)与MIP和NIP的选择性吸附结果(B)Fig.5 Structural formula of atrazine,mesotrione and sulcotrione(A)and their selective adsorption results by MIP and NIP(B)

式中,Q和C分别代表聚合物的吸附量以及溶液原浓度,δ1和δ2分别代表硝磺草酮和莠去津的静态分布系数,α1和α2为聚合物的选择性系数。
如表1 所示,MIP 的相对选择因子β为2.510,表明成功制备了选择性较高的聚合物。这是由于MIP 中的互补孔穴可以特异性识别模板分子。但莠去津与孔穴不能形成互补,所以其吸附量在MIP 和NIP上无明显区别。

表1 MIP和NIP选择性系数比较Table 1 Comparison of MIP and NIP selectivity coefficients
2.5 基质固相分散条件的选择
2.5.1 样品比例的选择实验研究了分散剂与样品的质量比(1∶2、2∶3、1∶1、3∶2、2∶1)对结果的影响。结果显示:分散剂与玉米样品质量比选择3∶2 和2∶1 时,硝磺草酮的回收率均较大(分别为95.8%、96.2%)。为节约分散剂用量,最终选择分散剂与样品的质量比为3∶2。
2.5.2 研磨时间的确定研磨可使分散剂与待提取样品充分接触,实验考察了不同研磨时间(2、5、10、15 min)时硝磺草酮的回收率。发现研磨10 min 时回收率最大(96.8%),继续延长研磨时间回收率无明显提高,因此实验选择研磨10 min。
2.5.3 淋洗剂、洗脱剂及其最佳体积的确定实验考察了5 种(水、20%乙腈水溶液、20%甲醇水溶液、50%乙腈水溶液、50%甲醇水溶液)淋洗剂对MIP 吸附选择性的影响。结果发现,纯水和20%甲醇水溶液得到的硝磺草酮回收率最大(分别为97.6%、97.3%),但纯水作为淋洗剂时,很难洗去杂质,故最佳淋洗剂选用20%甲醇水溶液。实验进一步考察了20%甲醇水溶液用量(1~7 mL)对结果的影响,发现2 mL用量时硝磺草酮的回收率最大。
实验考察了甲醇、乙酸、乙腈、1%乙酸乙腈、2%乙酸乙腈、5%乙酸乙腈为洗脱剂时对检测的影响。结果发现:5%乙酸乙腈溶液的洗脱效果最佳(96.3%),且其用量为1~5 mL时,回收率升高较快,用量为5~7 mL时,回收率保持较高值且趋于稳定。为节约试剂,最佳洗脱剂体积选择5 mL。
2.6 分析方法的评价与应用
在最佳检测条件下,得到硝磺草酮的回归方程为:A=111 872c-1 110.7(A为峰面积;c为硝磺草酮的含量,µg/g),r2=0.999 8,线性范围为0.05~10µg/g,检出限(S/N=3)为0.018µg/g。根据食品安全国家标准,玉米中硝磺草酮的最大残留值为0.5 mg/kg[19],所以本方法符合检测要求。为进一步评估方法准确度和精密度,使用本方法对加标玉米样品进行检测,得到回收率为97.0%~98.4%,相对标准偏差(RSD)为0.70%~5.6%。其加标样品的色谱图如图6所示。
采用本方法测定哈尔滨当地超市市售3 种玉米中的硝磺草酮含量。结果显示,3 种样品中均未检出硝磺草酮,随后对玉米样品在3 个水平下(0.05、0.30、5.00 µg/g)进行加标回收实验,得其回收率为97.5%~98.6%,RSD 为2.5%~4.8%。因此本实验所采用的检测方法在分离分析硝磺草酮方面具有一定的应用价值。
3 结 论
本文以硝磺草酮为模板分子,木质素为稳定粒子,采用Pickering 双乳液法制备了MIP,对其进行静态吸附、动态吸附、选择性吸附性能研究,并将其作为MSPD 的分散剂,萃取玉米中的硝磺草酮。结果显示:MIP 对硝磺草酮具有良好吸附作用,且动态吸附过程符合准二级动力学反应模型。本实验通过使用分子印迹聚合及基质固相分散技术,提高了分析检测效率,使木质素得到有效利用,有很好的实际应用价值。
猜你喜欢
陶瓷研究(2022年3期)2022-08-19
云南画报(2021年10期)2021-11-24
世界农药(2021年8期)2021-09-02
上海包装(2019年8期)2019-11-11
山东化工(2019年8期)2019-02-16
小学生优秀作文(高年级)(2018年4期)2018-09-11
现代农药(2018年1期)2018-03-01
天津造纸(2016年1期)2017-01-15
中国造纸学报(2015年1期)2015-12-16
中国摄影(2014年12期)2015-01-27
