
补体1q/肿瘤坏死因子相关蛋白9 改善急性心肌梗死小鼠心脏功能和减轻心肌组织纤维化
2022-01-09 06:31:24李香敏谭延振易定华
中国体外循环杂志 2021年6期
李香敏,谭延振,张 冰,赵 荣,易定华,孙 阳,易 蔚,张 平
急性心肌梗死(acute myocardial infarction,AMI)的发病率显著下降[1],随着早期再灌注治疗策略的实施,以及新的药理学方法的引入,AMI 的30天死亡率降低了60%,心脏急性事件期间预后获得了很大改善,但生存率的提高导致AMI 后慢性心力衰竭的患者明显增加[2-3]。 AMI 后心力衰竭的发病过程中抗心肌纤维化研究探索一直是心血管领域研究的热点,但是尚未研究出确实有效的治疗靶点和方法。 补体1q/肿瘤坏死因子相关蛋白9(complement 1q/ tumor necrosis factor related protein 9,CTRP9)作为一种分泌糖蛋白在心脏高度表达,参与多种代谢紊乱的发生和发展,在心血管疾病的保护、炎症的调控及糖脂代谢的调节中发挥重要的作用。CTRP9 在改善AMI 后心肌重塑中也发挥重要作用,这与其抗心肌纤维化密切相关,但具体的分子机制尚未完全明确,限制其进一步深入研究。
1 材料与方法
1.1 材料 八周龄健康雄性野生型C57BL/6 小鼠,由空军军医大学实验动物中心提供;CTRP9-KO 小鼠由南京大学生物医学研究中心构建。 实验动物的饲养及实验条件严格按照国家科学技术委员会颁布的《实验动物管理条例》进行。 抗转化生长因子(transforming growth factor,TGF)β1、抗波形蛋白(vimentin)抗体购于abcam 公司,苯基吲哚(DAPI)蓝色荧光购于LEAGENE 公司,C1qTNF9 酶联免疫吸附分析试剂盒购于Cloud-clone 公司,小鼠甘油醛-3-磷酸脱氢酶(glyceraldehyde-3-phosphate dehydrogenase,GAPDH)单克隆抗体购于CMCTAG 公司,小鼠TGF-β1 酶免试剂盒购于Elabscience 公司,H-89(二氢氯化物dihydrochloride)购于MedChemExpress公司,Phospho-PKA C(Thr197) (D45D3) 兔单克隆抗体购于CST 公司,重组补体1q/肿瘤坏死因子相关蛋白9(recombinant Complement 1q/ tumor necrosis factor related protein 9,rCTRP9)由本实验室合成。
1.2 方法
1.2.1 小鼠AMI 模型的构建 小鼠AMI 模型由美国Thomas Jefferson 大学的Gao Erhe 教授帮助构建。
1.2.2 动物实验分组及给药方式 手术4 h 后,待动物麻醉完全复苏后,将AMI 组C57BL/6 小鼠随机分为3 组:AMI+Vehicle 组(小鼠进行AMI 手术+腹腔注射生理盐水);AMI+CTRP9 组[小鼠进行AMI手术+腹腔注射rCTRP9 0.25 μg/(g·day)];AMI+CTRP9+H89 组[小鼠进行AMI 手术+腹腔注射H-89 0.5 μg/(g·day)+腹腔注射rCTRP9 0.25 μg/(g·day)];外加另外3 组:KO-AMI+Vehicle 组(KO 小鼠进行AMI 手术+腹腔注射生理盐水);野生型小鼠(wild type,WT)假手术(Sham)+Vehicle 组(WT 小鼠进行假手术操作+腹腔注射生理盐水);KO-Sham+Vehicle 组(KO 小鼠进行假手术操作+腹腔注射生理盐水)。
1.2.3 动物饲养管理 开放管理,自由采食,足量饮水。 每日检查有无死亡动物,AMI 后第4 天处死少量动物留取血清标本,手术后第28 天行超声心动图检测后,处死动物,留取标本。
1.2.4 心脏功能评估 应用Matrx 动物麻醉机将异氟烷和氧气混合后在麻醉盒内将小鼠麻醉后四肢用胶布固定并继续吸入混合气维持麻醉,使用Vevo2100 小动物超声影像系统检测并计算小鼠左心室射血分数(left ventricular ejection fraction,LVEF)。
1.2.5 取材 应用Matrx 动物麻醉机将异氟烷和氧气混合后在麻醉盒内将小鼠麻醉,称重、取血标本、取出心脏用磷酸盐缓冲液将血液冲洗干净后称重。
1.2.6 石蜡包埋小鼠心脏及切片 将小鼠左心室用多聚甲醛固定后修剪、石蜡包埋、切片。
1.2.7 小鼠心脏组织masson 染色 脱蜡至水、重铬酸钾染色、铁苏木素染色、丽春红酸性品红染色、磷钼酸染色、苯胺蓝染色、分化、透明封片。 结果判读:胶原纤维呈蓝色;肌纤维呈红色。
1.2.8 心肌组织免疫荧光染色检测CTRP9 表达及α-平滑肌肌动蛋白(α-smooth muscle actin,α-SMA)/vimentin 双染检测肌成纤维细胞 石蜡切片脱蜡至水、抗原修复、透化处理、封闭、孵化一抗(CTRP9 稀释比例为1 ∶50,α-SMA 稀释比例为1 ∶100,vimentin 稀释比例为1 ∶100)、孵化二抗、染核、封片,激光共聚焦显微镜观察。
1.2.9 波形蛋白vimentin 酶联免疫吸附试验(enzyme-linked immunosorbent assay,ELISA)检测小鼠血清CTRP9、TGF-β1 水平 参照ELISA 试剂盒步骤检测血清CTRP9、TGF-β1 水平变化。
1.2.10 免疫蛋白印迹(Western Blotting)检测心肌组织蛋白表达 Western Blotting 检测心肌组织胶原蛋白Ⅰ、TGF-β1、蛋白激酶A(proteinkinase A,PKA)、p-PKA 蛋白表达。
1.3 统计分析 文中和图中的所有数值均以n 个独立实验的均值±标准误(standard error of mean,SEM)表示。 数据用Graph Pad Prism-5 统计软件进行分析。 生存曲线采用Log-rank (Mantel-Cox)检验。 对两组数据采用t检验,对多组数据首先对所有实验组进行单因素方差分析,然后进行Bonferroni's 多重比较检验。P≤0.05(双侧)认为具有统计学意义。
2 结 果
2.1 AMI 28 d 后,小鼠生存率显著减低,心脏收缩功能明显降低,心肌纤维化程度明显增加 AMI+Vehicle 组死亡率为(10/27)、 Sham+Vehicle 组为(0/16), Log-rank (Mantel-Cox)检验,I2=7.233,P=0.0072,AMI 后小鼠生存显著降低;Sham+Vehicle组心脏重量/体重为(4.475±0.165)mg/g, AMI+Vehicle 组心脏重量/体重为(6.675±0.135)mg/g,AMI后小鼠心脏变大;Sham+Vehicle 组LVEF 为(74.460±1.668)%,AMI+Vehicle 组LVEF 为(25.190±1.488)%,AMI 后小鼠心脏收缩功能明显减低;Sham+Vehicle组(左室游离壁)纤维化为(0.495±0.084)%,AMI+Vehicle 组(梗死交界区)心肌纤维化为(25.410±1.041)%,AMI 后小鼠梗死交界区心肌纤维化明显增加。 见图1。
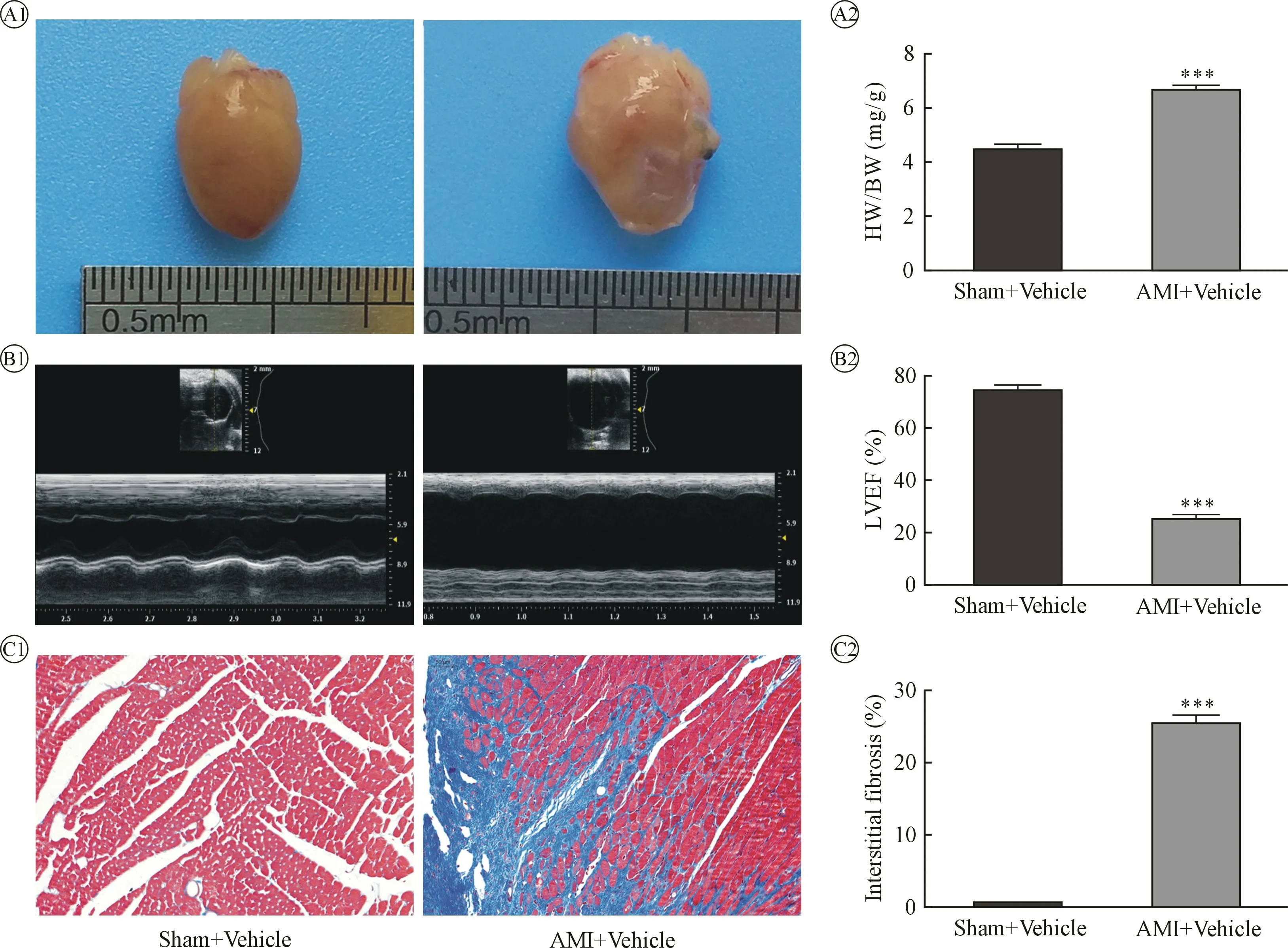
图1 Sham+Vehicle 组与AMI+Vehicle 组心脏大小、收缩功能、纤维化程度比较
2.2 AMI 后小鼠心肌组织及血清CTRP9 水平降低,血清TGF-β1 水平明显升高,梗死交界区肌成纤维细胞数量明显增加,PKA 活性增强,纤维化相关蛋白表达明显增加 小鼠AMI 后第28 天应用免疫荧光染色检测小鼠AMI 后心肌组织CTRP9 表达变化,与Sham+Vehicle 组(左室游离壁)比较,AMI+Vehicle 组CTRP9 在远离区、交界区及梗死区表达均有不同程度降低,且越接近梗死区,降低越严重。 见图2。
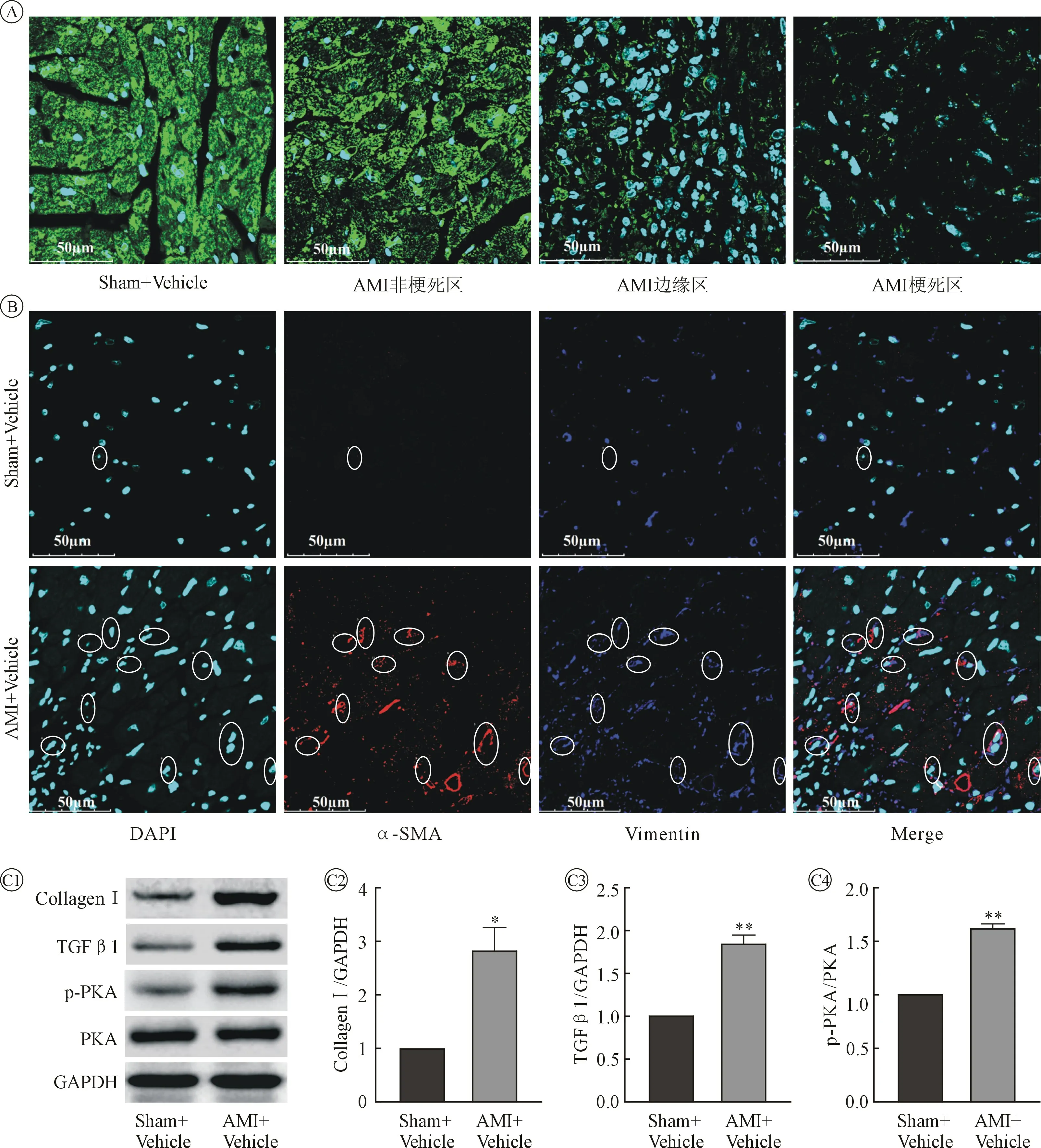
图2 Sham+Vehicle 组与AMI+Vehicle 组心肌组织染色图像及蛋白变化比较
小鼠AMI 后第4 天Sham+Vehicle 组血清CTRP9 为(907.000±40.190) μg/L, AMI+Vehicle 组为(519.700±17.770)μg/L,n =5,t检验P<0.001,AMI 后小鼠血清CTRP9 水平明显降低。 小鼠AMI后第4 天Sham+Vehicle 组血清TGF-β1 为(66.790±2.690)ng/L, AMI+Vehicle 组为(118.500±6.872)ng/L,n =5,t检验P<0.001,AMI 后小鼠血清TGFβ1 水平明显升高。
小鼠AMI 后第28 天Sham+ Vehicle 组心肌组织中(左室游离壁)肌成纤维细胞数量(α-SMA/Vimentin 双染阳性)为(0.250±0.250),AMI+Vehicle 组(梗死交界区)为(7.500±0.646),n =4,t检验P<0.001,AMI 后小鼠梗死交界区肌成纤维细胞数量增加。
小鼠AMI 后第28 天AMI+ Vehicle 组心肌组织(取自梗死交界区)胶原蛋白Ⅰ(Collagen Ⅰ)、TGFβ1、p-PKA/PKA 分别为Sham+Vehicle 组(取左室游离壁)的(2.819±0.427)倍、(1.839±0.089)倍、(1.605±0.062)倍,n =4,t检验P=0.0238、P=0.0025、P=0.0023,AMI 后胶原蛋白Ⅰ、TGF-β1 表达升高,PKA磷酸化水平增高,活性显著增加。
2.3 敲除CTRP9 AMI 小鼠较野生型AMI 小鼠28天死亡率更高,心脏收缩功能更差,心肌纤维化更加严重,PKA 活性受到部分抑制,纤维化相关蛋白表达增加更多 WT-Sham+Vehicle 组小鼠第28 天死亡率为(0/13),KO-Sham+Vehicle 组为(0/11),WT-AMI+Vehicle 组为(10/27),KO-AMI+Vehicle 组为(10/22)。 4 组Log-rank (Mantel-Cox)检验,χ2=12.52,P=0.0058,4 组之间比较有显著的统计学意义,虽然KO-AMI+ Vehicle 组较WT-AMI+Vehicle组死亡率增加,但2 组之间χ2=0.2983,P=0.5849,生存曲线未达到统计差异。 见图3。

图3 WT-Sham+Vehicle、KO-Sham+Vehicle、WT-AMI+Vehicle、KO-AMI+Vehicle 四组检测图像和各项统计比较
WT-Sham+Vehicle 组心脏重量/体重为(4.475±0.165)mg/g,n =6,KO-Sham+Vehicle 组心脏重量/体重为(4.633±0.194)mg/g,n =6,t检验P=0.5485,KO-CTRP9+Vehicle 组小鼠心脏大小与WT-Sham+Vehicle 组小鼠无差异;WT-AMI+Vehicle 组心脏重量/体重为(6.675±0.135)mg/g,n =6,KO- AMI+Vehicle 组心脏重量/体重为(7.233 ±0.145)mg/g,n =6,t检验P=0.0182, AMI 后KO-CTRP9 型小鼠心脏比WT-AMI 组明显增大。
WT-Sham+Vehicle 组LVEF 为(74.460±1.668)%,n =5,KO-Sham+Vehicle 组LVEF 为(73.400±1.839)%,n=5,t检验P=0.7931,两组小鼠心脏收缩功能无差异;KO-AMI+Vehicle 组LVEF 为(15.100±1.345)%,n=5,WT-AMI+Vehicle 组LVEF 为(25.190±1.488)%,n =5,t检验P=0.0010,KO-AMI+Vehicle 组小鼠心脏收缩功能减低更严重。
Masson 染色检测KO-Sham+Vehicle 组(左室游离壁)纤维化为(0.495±0.084)%,n =4,KO-Sham+Vehicle 组(梗死交界区)纤维化为(0.510±0.103)%,n =4,t检验P=0.9139,两组小鼠心肌纤维化程度无差异;WT-AMI+Vehicle 组纤维化为(25.410±1.041)%,n =4,KO-AMI+Vehicle 组纤维化为(31.550±2.098)%,n =4,t检验P=0.0395, KO-AMI+Vehicle 组小鼠心脏纤维化较WT-AMI+Vehicle 组小鼠严重。
KO-Sham+ Vehicle 组心肌组织胶原蛋白Ⅰ、TGF-β1、p-PKA/PKA 分别为WT-Sham+ Vehicle组的(1.029±0.079)倍、(1.010±0.088)倍、(0.984±0.080)倍,n =4,t检验P=0.7365、P=0.9197、P=0.8494,两组之间胶原蛋白Ⅰ、TGF-β1、PKA 磷酸化水平无差异;WT-AMI+Vehicle 组心肌组织胶原蛋白Ⅰ、TGF-β1、p-PKA/PKA 分别为WT-Sham+Vehicle 组的(2.819±0.427)倍、(1.839±0.089)倍、(1.605±0.062)倍,KO- AMI+Vehicle 组心肌组织胶原蛋白Ⅰ、TGF-β1、p-PKA/PKA 分别为WT-Sham+Vehicle 组的(3.110±0.464)倍、(2.257±0.092)倍、(1.465±0.047)倍,n =4,WT-AMI+Vehicle 组与KO-AMI+Vehicle 组之间进行t检验,P=0.0090、P=0.0126、P=0.0063,KO-AMI+Vehicle 组较WT-AMI+Vehicle 组胶原蛋白Ⅰ、TGF-β1 表达升高,PKA 磷酸化水平减低,活性受到抑制。
2.4 补充rCTRP9 后小鼠AMI 第28 天的死亡率较AMI+Vehicle 组降低,心脏收缩功能改善,心肌纤维化减轻,纤维化相关蛋白表达减少,且这种作用可被PKA 抑制剂H-89 阻断 AMI 后第28 天AMI+Vehicle 组小鼠死亡率为(10/27), AMI+CTRP9 组为(4/26),AMI+CTRP9+H-89 组为(9/27);Log-rank(Mantel-Cox)检验,χ2=3.272,P=0.1948。 虽然AMI+CTRP9 组死亡率较AMI+Vehicle 组及AMI+CTRP9+H-89 组减低,但三组生存曲线未达到统计差异。 见图4。
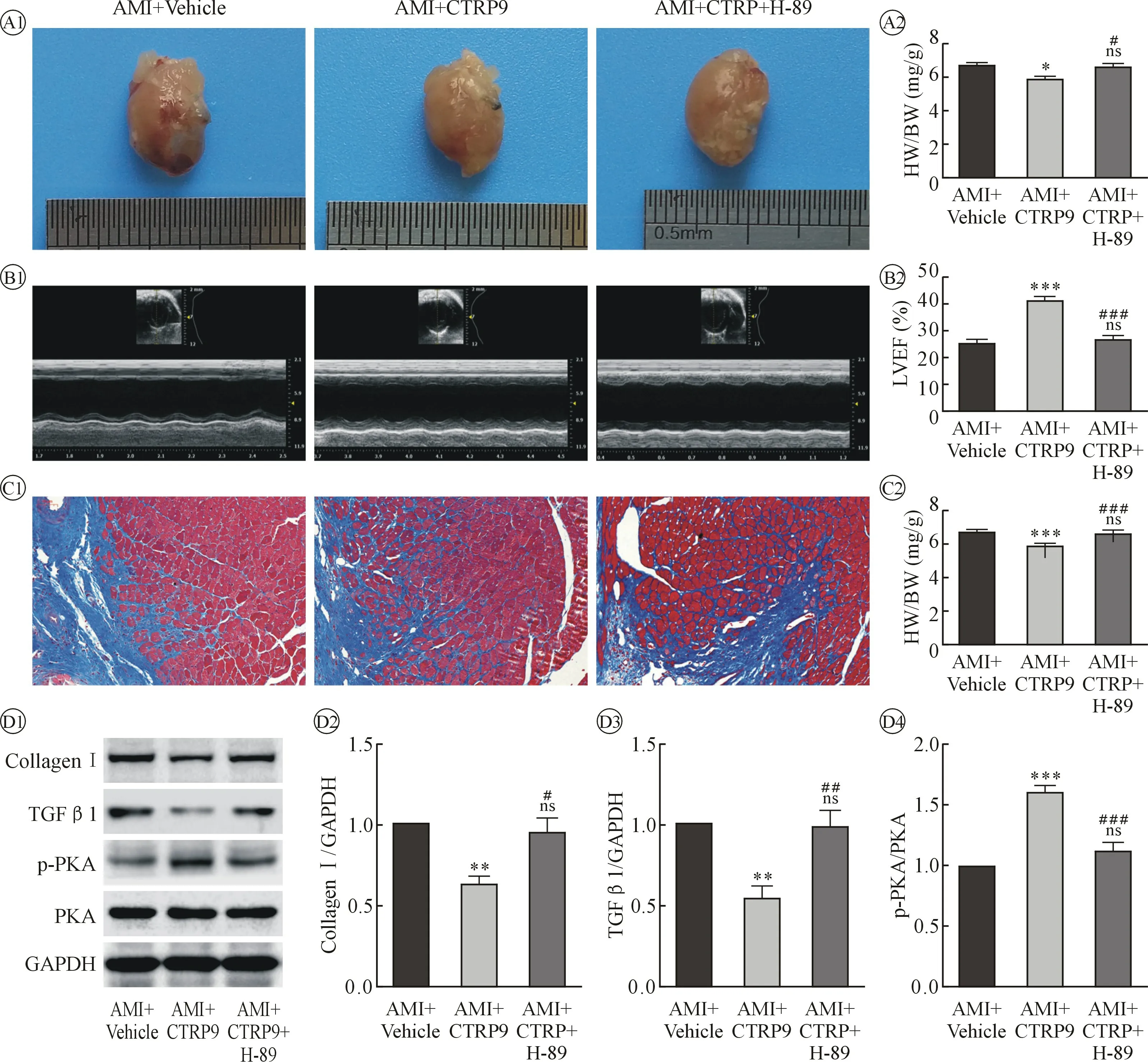
图4 AMI+ Vehicle、AMI+CTRP9、AMI+CTRP9+H-89 三组检测图像和各项统计比较
AMI+Vehicle 组心脏重量/体重为(6.675±0.135)mg/g,n =6,AMI+CTRP9 组心脏重量/体重为(5.833±0.182)mg/g,n =6,AMI+CTRP9+H-89 组心脏重量/体重为(6.600±0.193)mg/g,n =6,AMI+CTRP9 组小鼠心脏比AMI+Vehicle 组小鼠减小,AMI+Vehicle 组和AMI+CTRP9+H-89 组小鼠心脏大小无差异,AMI+CTRP9+H-89 组较AMI+CTRP9 组小鼠心脏增大。
心脏超声检查AMI+Vehicle 组LVEF 为(25.190±1.488)%,AMI+CTRP9 组LVEF 为(41.250±1.493)%,AMI+CTRP9+H-89 组LVEF 为(26.760±1.481)%,n =5,AMI+CTRP9 组小鼠心脏收缩功能较AMI+Vehicle 组明显增强,AMI+Vehicle 组和AMI+CTRP9+H-89 组小鼠心脏收缩功能无差异,AMI+CTRP9+H-89 组较AMI+CTRP9 组小鼠心脏收缩功能明显差。
Masson 染色检测AMI+Vehicle 组(梗死交界区)纤维化为(25.410 ±1.041)%,AMI+CTRP9 组纤维化为(17.510 ±0.893)%,n =4,AMI+CTRP9+H-89 组纤维化为(25.430 ±1.031)%,n =4,AMI+CTRP9 组小鼠心肌纤维化程度较AMI+Vehicle 组明显减轻,AMI+Vehicle 组和AMI+CTRP9+H-89 组小鼠心肌纤维化程度无差异,AMI+CTRP9+H-89 组较AMI+CTRP9 组小鼠心肌纤维化程度明显重。
Western Blotting 检测AMI+CTRP9 组心肌组织(梗死交界区)蛋白表达变化原蛋白Ⅰ、TGF-β1、p-PKA/PKA 分别为AMI+Vehicle 组的(0.624±0.054)倍、(0.538±0.082)倍、(1.598±0.058)倍,AMI+CTRP9+H-89 组心肌组织(梗死交界区)蛋白表达变化原蛋白Ⅰ、TGF-β1、p-PKA/PKA 分别为AMI+Vehicle 组的(0.947±0.096)倍、(0.988 ± 0.101)倍、(1.113±0.078)倍,n =4,AMI+CTRP9 组小鼠心肌组织胶原蛋白Ⅰ、TGF-β1 的表达量较AMI +Vehicle明显减少,AMI+Vehicle 组和AMI+CTRP9+H-89 组小鼠心肌组织胶原蛋白Ⅰ、TGF-β1 的表达量无差异,AMI+CTRP9+H-89 组较AMI+CTRP9 组小鼠心肌组织胶原蛋白Ⅰ、TGF-β1 的表达量明显增加,AMI+CTRP9 组小鼠PKA 磷酸化激活程度较AMI+Vehicle 升高,AMI+Vehicle 组和AMI+CTRP9+H-89组小鼠PKA 磷酸化激活程度相同,AMI+CTRP9+H-89 组较AMI+CTRP9 组小鼠PKA 磷酸化激活减低。
3 讨 论
缺血性心脏病在全球范围内仍然是主要死亡原因[4],心肌成纤维细胞通过沉积细胞外基质来修复坏死心肌组织,在心力衰竭的发病机制中起着关键作用[5]。 AMI 后非梗死区局部结缔组织的扩张,梗死边缘区和远离区未受损伤心肌的反应性纤维化导致心肌硬度增加,顺应性降低,从而进一步降低心输出量[6],引起心脏功能衰竭,这是造成AMI 致死的重要原因之一。
CTRP9 是一种在心脏高度表达的分泌糖蛋白,在心血管疾病的保护、炎症的调控及糖脂代谢的调节中发挥重要的作用,本研究组在过去研究中发现在缺血性心脏损伤中,血浆和心室组织中CTRP9 的表达显著降低,CTRP9 具有明显的心肌保护作用,CTRP9 通过改善心肌纤维化在改善心肌重塑中发挥重要作用[7]。 但目前研究CTRP9 主要以心肌细胞、血管内皮细胞为研究对象,以心肌成纤维细胞作为对象的研究尚未见报道。
本研究应用免疫荧光染色的方法直接观察了AMI 不同区域CTRP9 的表达变化,与缺血的严重程度直接相关,表达水平从梗死远离区、交界区、梗死区依次明显降低。 在小鼠AMI 后第4 天血清中CTRP9 水平明显减低伴随着血清中TGF-β1 明显升高。 TGF-β 家族是研究最多的纤维反应调节因子之一,它在心肌损伤后的适应和重塑中起着重要作用[8-10]。 本研究发现AMI 后28 天梗死交界区肌成纤维细胞数量明显增加。 肌成纤维细胞分泌大量结构细胞外基质蛋白[11-12],可以引起心肌组织纤维化,在梗死交界区纤维化明显增加。 这些结果提示在AMI 过程中CTRP9 的表达降低及TGF-β1 的升高可引起成纤维细胞的持续激活,分泌大量基质蛋白造成心肌组织发生纤维化。
为了进一步证实这一观点,本研究构建了CPRT9 敲除小鼠,在无AMI 时,KO-CTRP9 小鼠与WT 型小鼠心肌纤维化程度及心脏收缩功能基本一致,但在AMI 发生后,KO-CTRP9 小鼠的心肌纤维化程度更加严重,心脏收缩功能降低更加严重,从反面证实CTRP9 的减低参与了AMI 后过度的心肌纤维化并且造成心脏收缩功能的进一步减低。 补充rCTRP9 后,心肌纤维化明显减轻,心脏收缩功能明显改善,从正面证实补充CTRP9 可减低AMI 后的心肌纤维化并且改善心脏的收缩功能。
前期研究证实CTRP9 可激活促生存信号分子PKA 而抑制H9C2 细胞缺氧损伤引起的心肌细胞凋亡减轻心肌重塑[7]。 本研究发现在AMI 后第28天,PKA 活性水平比对照组明显升高,而KOCTRP9 小鼠的AMI 发生后,PKA 活性水平比野生型AMI 小鼠明显减低;补充rCTRP9 后PKA 活性水平进一步明显提高,从正反两面证实在整体水平CTRP9 通过影响PKA 活性发挥抗AMI 后心肌纤维化,改善心脏收缩功能。
为了进一步研究PKA 通路在CTRP9 抗AMI 后心肌纤维化中的作用,在动物水平应用PKA 抑制剂H-89 阻断PKA 活性升高,观察CTRP9 抗AMI 后心肌纤维化的作用的变化。 发现应用H-89 后补充rCTRP9 引起的PKA 活性水平提高被阻断,同时Masson 染色检测AMI 交界区纤维化结果显示CTRP9 抗AMI 后心肌纤维化作用也被阻断,CTRP9改善心脏收缩功能的作用也随之被阻断。
本研究证实CTRP9 有通过PKA 通路减轻AMI小鼠心肌组织纤维化和改善心脏功能的作用。
猜你喜欢
传染病信息(2022年3期)2022-07-15 08:24:28
法人(2021年12期)2021-05-09 17:24:24
肝博士(2021年1期)2021-03-29 02:32:16
Coco薇(2017年12期)2018-01-03 21:27:09
天然产物研究与开发(2016年6期)2016-06-05 10:29:30
现代食品(2016年14期)2016-04-28 08:10:07
广东海洋大学学报(2015年4期)2016-01-13 08:39:40
医学研究杂志(2015年6期)2015-07-01 17:40:08
中国中医药现代远程教育(2014年13期)2014-03-01 04:26:57
河南医学研究(2014年3期)2014-02-27 14:51:51
