
高效液相色谱双波长法同时测定复方枇杷喷托维林颗粒中三种成分
2022-01-04 08:09:50任静杜晖
安徽医药 2022年1期
任静,杜晖
复方枇杷喷托维林颗粒是由枇杷叶流浸膏、甘草流浸膏和枸橼酸喷托维林等按一定比例制成,可用于上呼吸道感染、支气管炎等引起的干咳或咳嗽少痰。枸橼酸喷托维林是制剂中的化学药组分,它是一种非成瘾性中枢镇咳药,实验中多采用HPLC法测定其含量[1]。甘草酸是甘草流浸膏中最主要的活性成分,包括其二铵盐、苷类具有抗炎[2-4]、抗病毒[5-7]、抗动脉粥样硬化[8-9]及抑制肝损伤[10-14]等药理作用;而甘草次酸是甘草酸在体内的代谢产物,具有肝脏保护[15-16]和抗氧化作用[17]。有报道表明甘草酸及其代谢产物是中药制剂发挥镇咳功效的“使药”成分之一[18],所以两者的含量与制剂的药理活性紧密相关。该药品现行标准中采用紫外分光光度法测定制剂中枸橼酸喷托维林的含量,方法需要经甲基橙显色氯仿提取,重现性较差[19-20];有文献对其中枸橼酸喷托维林和甘草酸分别进行含量测定[20-21],但因采用两种完全不同的色谱体系导致测定烦琐,也无甘草酸代谢产物甘草次酸的含量控制项目。因此,建立一种能够同时测定多种成分含量的方法尤为重要。本研究采用HPLC双波长法梯度洗脱快速测定复方枇杷喷托维林颗粒中枸橼酸喷托维林、甘草酸和甘草次酸三种成分的含量,为该制剂质量标准的完善和提高提供参考。本研究起止时间为2019年6月至2020年5月,符合《世界医学协会赫尔辛基宣言》相关要求。
1 材料与方法
1.1 仪器与试药热电Ultimate 3000高效液相色谱仪(美国赛默飞世尔科技公司);Milli-Q Advantage超纯水机(美国密理博公司);MS105十万分之一天平(瑞士梅特勒公司)。
复方枇杷喷托维林颗粒(江西赣南海欣药业股份有限公司,批号15121001、15121002、15012203);甘草酸铵(批号110731~201720,含量97.7%)、甘草次酸(批号110723~201715,含量99.6%)和枸橼酸喷托维林(批号100432~201803,含量99.8%)等对照品均来自中国食品药品检定研究院;色谱纯甲醇来自美国霍尼韦尔公司;实验用水由Milli-Q系统制备。
1.2 方法
1.2.1 溶液的制备
1.2.1.1 样品溶液的制备取10袋颗粒混合,研细,称取适量(约10.000 g),置100 mL量瓶中,加50 mL水振摇使溶解,再用甲醇定容,滤过,取续滤液经0.45 μm微孔滤膜过滤,即得。
1.2.1.2 混合对照品溶液的制备精密称取甘草次酸对照品10.09 mg,置20 mL量瓶中,加甲醇适量,振摇使溶解,用甲醇稀释至刻度;精密称取枸橼酸喷托维林对照品20.52 mg和甘草酸铵对照品10.02 mg,置20 mL量瓶中,加甲醇适量,振摇使溶解,精密加入甘草次酸对照品溶液2 mL,用甲醇稀释至刻度,作为混合对照品储备溶液。分别精密量取0.2 mL、0.4 mL、0.8 mL、2 mL、4 mL和10 mL,置20 mL量瓶中,用甲醇稀释至刻度,即为混合对照品溶液。
1.2.2 色谱条件热电ScientificTMAcclaimTM120Å C18色谱柱(250 mm×4.6 mm,5 μm);流动相A为甲醇,B为1%三乙胺水溶液(用磷酸调pH为3.0),二元梯度洗脱(0~5 min,60%A;5~20 min,A相线性上升至90%;20~25 min,90%A;25.1~30 min,60%A);流速1.0 mL∕min;柱温30℃;进样量10 μL;DAD检测(波长设置为215 nm和250 nm)。
2 结果
2.1 线性关系考察精密量取“1.2.1.2”项下配制的枸橼酸喷托维林、甘草酸和甘草次酸的混合对照品溶液,分别注入色谱仪,以主成分峰面积Y(mAU·min)对浓度X(μg∕mL)进行线性回归,相关系数(r)均大于0.999 9,见表1。
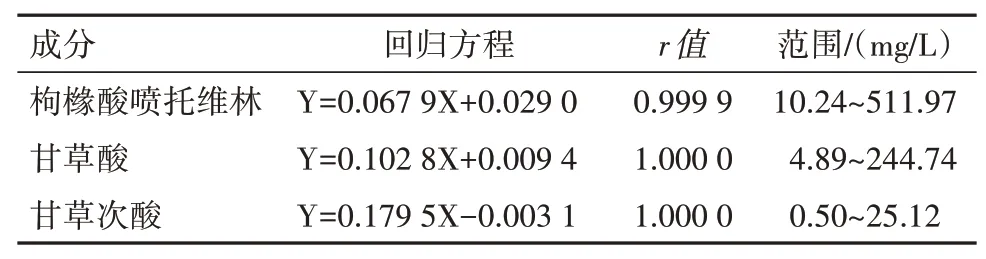
表1 三种组分的回归方程、相关系数和线性范围
2.2 专属性及样品含量测定分别吸取混合对照品、批号为15012203的供试品和阴性样品溶液10 μL,注入色谱仪,记录色谱图如图1所示,枸橼酸喷托维林、甘草酸和甘草次酸的保留时间分别为6.5 min、14.1 min和23.3 min,三种成分与杂质之间分离良好。
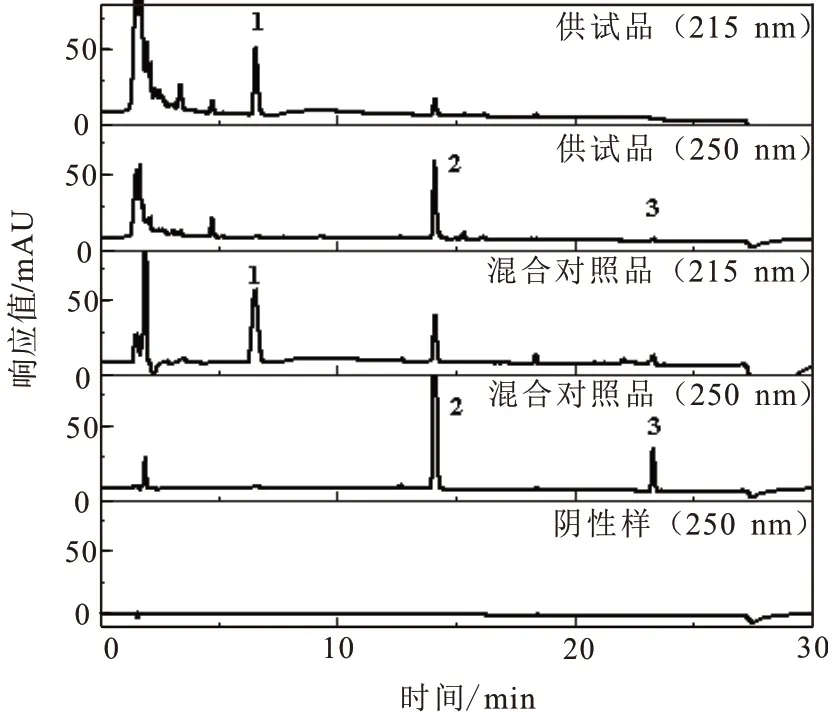
图1 HPLC色谱图
按“1.2.2”项色谱条件测定三批复方枇杷喷托维林颗粒中三种组分的含量,如表2所示,枸橼酸喷托维林和甘草酸的含量与处方量和文献值基本一致[20-21]。其中甘草酸含量较之甘草流浸膏中甘草酸限值略低[22],可能是颗粒长时间存放或运输途中发生了变化。
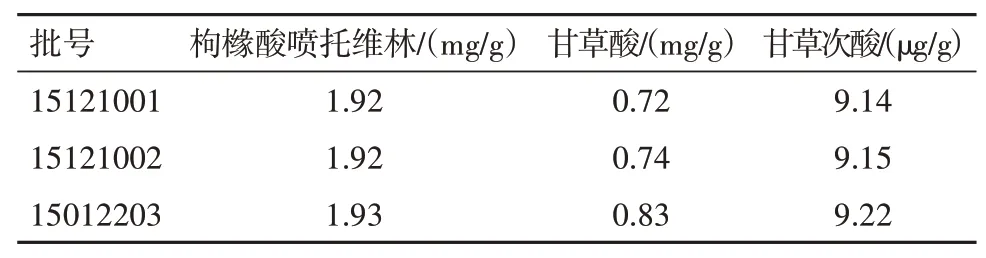
表2 样品测定结果
2.3 精密度考察将批号为15012203的样品溶液连续进样6次,发现枸橼酸喷托维林、甘草酸和甘草次酸峰面积的RSD值分别为0.60%、0.59%和0.38%,证明仪器精密度能满足实验要求。
平行处理6份批号为15012203的样品,按2.2项色谱条件测定样品中枸橼酸喷托维林、甘草酸和甘草次酸的含量,RSD值分别为0.68%、0.25%和0.79%,方法重复性良好。
为了考察不同人员和仪器对精密度的影响,重新配制混合对照品溶液并处理6份批号为15012203的样品,测得样品中枸橼酸喷托维林、甘草酸和甘草次酸的平均含量分别为1.88 mg∕g,0.80 mg∕g和9.18 μg∕g,对 应 的RSD分 别 为1.25%、0.52%和0.91%。结果显示,枸橼酸喷托维林、甘草酸和甘草次酸的F值分别为3.21、4.02和1.32,均小于置信度为95%时的F(5,5)值(5.05),说明两组精密度结果差异无统计学意义。
2.4 稳定性考察将上述溶液保存在4℃环境中,并在6 h、12 h、24 h和48 h后进样分析,三种组分含量的RSD值分别为1.51%、0.89%和2.02%,说明样品溶液在2 d内保持稳定。
2.5 回收率考察精密称取甘草次酸对照品4.52 mg,置100 mL量瓶中,用甲醇溶解并稀释至刻度,作为甘草次酸溶液。精密称取枸橼酸喷托维林对照品101.70 mg和甘草酸铵对照品41.45 mg,置同一10 mL量瓶中,用上述溶液溶解并稀释至刻度。精密称取批号为15012203的样品约5.00 g,置100 mL量瓶中,平行9份,分别精密加入上述混合对照品溶液0.50 mL、1.00 mL和1.50 mL,每组平行3份,按“1.2.1.1”项下方法对样品进行处理,计算平均回收率,枸橼酸喷托维林、甘草酸和甘草次酸的平均回收率分别为102.27%,99.94%和98.02%,相应的RSD为2.31%,2.33%和2.42%,结果如表3所示。
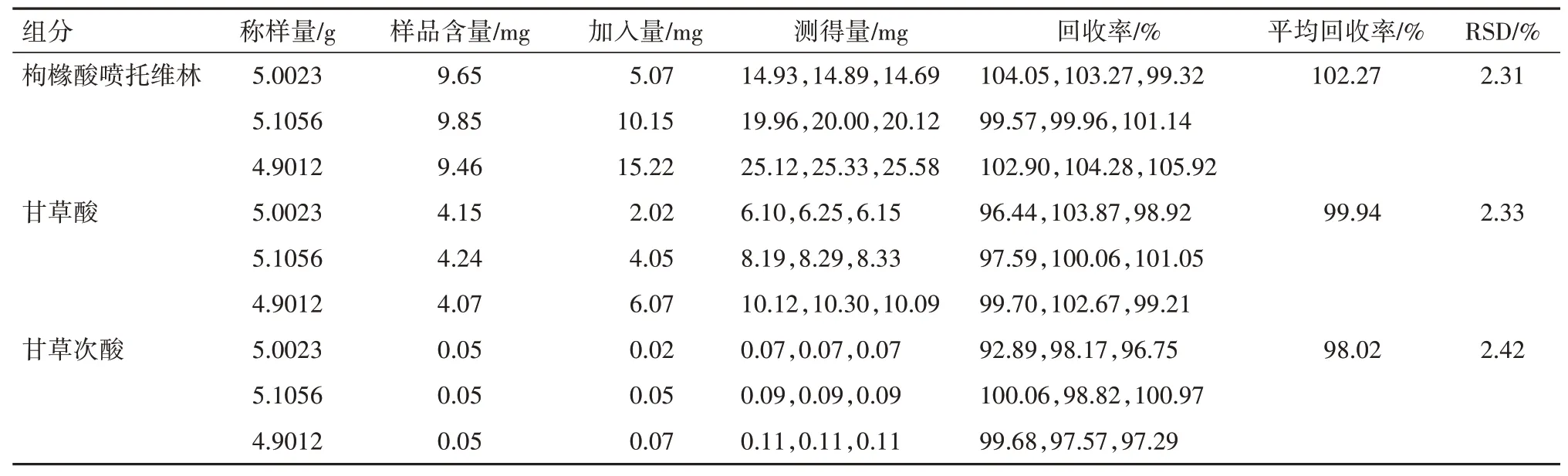
表3 三种组分的回收率
3 讨论
检测波长的选择:枸橼酸喷托维林的最大吸收波长为200 nm,而甘草酸和甘草次酸的最大吸收为250 nm。甲醇的紫外吸收波长为205 nm,在此范围内测定基线不平稳,干扰较明显;但波长大于215 nm,喷托维林的紫外吸收又明显减弱。综合考虑,我们设置为双波长215 nm和250 nm同时检测。
提取溶剂的选择:比较了相同的超声时间下,甲醇、甲醇-水(50∶50)和水等三种溶剂的提取率和样品出峰情况。试验发现,甲醇的提取率最低,而颗粒在水中的溶解性最好;但溶剂与流动相极性差别较大导致目标物的色谱峰延展严重。故我们在水提物中加入相同比例的甲醇,从而满足我们的分析要求。
流动相的选择:本组尝试采用乙腈-水(60∶40)和甲醇-水(70∶30)等度洗脱,发现甘草酸和甘草次酸的出峰时间较晚,且色谱峰有拖尾现象,故在水相中加入1%的三乙胺用以改善峰形。我们发现喷托维林和甘草酸的色谱峰在乙腈体系中较之甲醇宽,塔板数略低;另外药典中对喷托维林的测定亦采用甲醇-水体系[23]。但是提高甲醇比例,枸橼酸喷托维林又与相邻杂质峰无法基线分离。最后我们降低初始状态下有机相比例,采用甲醇-1%三乙胺水溶液(用磷酸调pH为3.0)梯度洗脱,分离度和重现性较好。
现行标准中采用甲基橙分光光度法测定喷托维林的含量,且缺乏甘草酸和甘草次酸等成分的测定项目,所以标准的专属性和全面性存在较大缺陷。本研究采用HPLC双波长法梯度洗脱同时对颗粒中三种组分进行分离测定,简化了文献中2个色谱体系才能完成的检验任务,大大缩短了分析时间,为该制剂质量标准的完善和提高提供了参考。
猜你喜欢
基层中医药(2022年7期)2022-11-17 08:25:08
中成药(2018年2期)2018-05-09 07:20:08
医药前沿(2018年2期)2018-01-17 12:10:56
中成药(2017年3期)2017-05-17 06:08:48
实用临床医学(2016年8期)2016-06-07 01:28:16
中国卫生标准管理(2015年4期)2016-01-14 05:16:45
山东医药(2015年16期)2016-01-12 00:40:04
西南医科大学学报(2016年4期)2016-01-03 01:26:29
中国当代医药(2015年33期)2015-03-01 02:09:17
中国药业(2014年20期)2014-05-17 03:13:44
