
CaMKII/NFAT通路激活可促进AKI小鼠肾小管上皮细胞凋亡*
2021-12-30 05:22黄宗顺彭用华俞小敏陈亚琦
中国病理生理杂志 2021年12期
黄宗顺, 肖 洁, 彭用华, 俞小敏, 陈亚琦
(广州医科大学附属第一医院肾内科,广东广州510120)
急性肾损伤(acute kidney injury,AKI)是肾功能在短期内下降而出现的氮质废物滞留和尿量减少的综合征。AKI 具有全球发病率高、预后不良和医疗费用高等严峻问题[1-6]。目前,AKI 的发病机制尚未明确。肾小管上皮细胞(renal tubular epithelial cells,RTEC)凋亡是AKI 发生发展的重要病理改变之一,明确RTEC 凋亡的分子机制对AKI的防治具有重要意义。钙/钙调蛋白依赖性蛋白激酶II(calcium/calmodulin-dependent protein kinase II,CaMKII)是广泛表达于各种组织的丝氨酸/苏氨酸蛋白激酶,与细胞凋亡密切相关[7-9]。CaMKII 在氧化应激损伤和细胞凋亡中发挥重要作用,敲除CaMKII基因或抑制其功能可缓解内质网应激诱发的心肌细胞凋亡[10]。也有研究认为,CaMKII 过表达可促进碘海醇诱导的线粒体损伤和 RTEC 凋亡[7]。目前,CaMKII 是否参与AKI 的发病及其在RTEC 凋亡中的机制仍未明确。因此,本研究采用阿霉素(adriamycin,ADR)干预人近端肾小管上皮细胞HK-2 和BALB/c 小鼠,分别建立 RTEC 凋亡细胞模型和 AKI 动物模型[11-12];采用Western blot、流式细胞术和细胞转染等技术,初步探讨CaMKII 和活化T 细胞核转录因子(nuclear factor of activated T cells,NFAT)在 AKI 的 RTEC 凋亡中的作用和调控机制。
材料和方法
1 实验材料
1.1 细胞培养与分组 肾小管上皮细胞HK-2(购自ATCC)用含10%胎牛血清(Gibco BRL)的RPMI-1640 培养液(Gibco BRL)在37 ℃培养箱中培养。培养成熟的RTEC 予ADR(0.062 5 mg/L;购自Sigma)、KN-93 磷酸盐(简写为K;CaMKII 的特异性抑制剂;2 μmol/L;购自Selleck Chemicals)、钙调蛋白(calmodulin,CaM;CaMKII 活化剂;5 μmol/L)或siRNA(50 nmol/L)干预24 h。培养成熟的RTEC 按实验内容随机分组。第1~3 部分实验分为3 组:正常对照(control,Con)组、ADR 组和 ADR+K 组。第 4 部分实验分为 6 组:Con 组、siRNA 空白对照(siRNA blank control,siCon)组、ADR 组、ADR+NFAT-siRNA(siNFAT)组和CaM 组和CaM+siNFAT 组。siNFAT(序列为 5′-GCCATAACTTTCTGCAAGA-3′)和siCon 购自广州锐博公司。siRNA 的细胞转染方法详见我们的前期研究[12-15]和产品说明书。
1.2 实验动物分组与干预 18 只 SPF 级 6~8 周龄18~21 g 的雌性BALB/c 小鼠购自北京维通利华实验动物技术有限公司,许可证号为SCXK(京)2016-0006。按每组 6 只小鼠随机分为 3 组:Con 组、ADR组和ADR+K 组。动物实验伦理通过广州医科大学附属第一医院伦理委员会的审查。12只小鼠在实验前 1 d 尾静脉注射 ADR(12 mg/kg),6 只 Con 组小鼠尾静脉注射相同体积的生理盐水。其中6 只注射ADR 的小鼠在实验第 1、3、5 天予腹腔注射 K(0.08 mg/kg)。因造模时小鼠意外死亡2只,最后入组情况为每组5 只。小鼠在第7 天予氯胺酮(70 mg/kg 腹腔注射)麻醉后处死,取动脉血提取血清行酶联免疫吸附实验,肾脏组织用于提取蛋白行免疫印迹实验。
2 方法
2.1 Western blot 具体步骤详见我们的以往研究[12-15]。简述如下:按照核浆蛋白提取试剂盒(南京凯基生物科技发展有限公司)说明提取总蛋白、核蛋白与胞浆蛋白。BCA 法测量各样品的蛋白浓度。蛋白变性后以每个电泳道上样30 μg 进行SDS-PAGE。转膜后剪下目的条带,用5%脱脂奶粉室温条件下封闭1 h 后,加相应Ⅰ抗4 ℃摇床孵育过夜。次日洗去Ⅰ抗后加Ⅱ抗室温摇床孵育1 h,用TBST 洗条带3次,每次5 min。ECL 发光液显影、拍照。实验所用Ⅰ抗体如下:兔抗p-CaMKII(Cell Signaling Technology,1∶1 000),兔抗t-CaMKII(Santa Cruz,1∶500),兔抗NFAT(Cell Signaling Technology,1∶1 000),兔抗Bcl-2(Cell Signaling Technology,1∶1 000),兔抗Bax(Cell Signaling Technology,1∶1 000),兔抗 GAPDH(Bioworld Technology,1∶10 000),兔抗 histone(Cell Signaling Technology,1∶3 000)。蛋白表达定量以目标蛋白/GAPDH或目标蛋白/histone(核蛋白)表示。
2.2 流式细胞术 具体实验步骤见我们以往研究[12-15],各组 RTEC 的细胞凋亡率严格按照 Annexin V/碘化丙啶(propidium iodide,PI)凋亡检测试剂盒(南京凯基生物科技发展有限公司)的说明执行。简述如下:以磷酸缓冲盐溶液洗净各组HK-2 细胞,0.05%胰酶消化细胞使其从培养皿脱落,用含5%胎牛血清的培养液终止消化。冷磷酸缓冲盐溶液洗净HK-2 细胞后离心管收集约5×105细胞。用结合缓冲液(Binding Buffer)重悬HK-2 后转移至流式管。先后加Annexin V-FITC(染凋亡细胞)和PI 混匀。室温避光条件下孵育10 min。最后在1 h 内用流式细胞仪检测各标本的细胞凋亡率。
2.3 酶联免疫吸附实验 各组小鼠的血清肌酐检测严格按照小鼠肌酐检测试剂盒(Cayman Chemical)的说明执行。简述如下:准备各梯度浓度的标准品。酶标板上设空白孔、标准孔和待测样品孔,加入各标准品或样品15 μL。每孔再加150 μL 碱性苦味酸溶液。室温摇床孵育10 min。用酶标仪492 nm 波长下测各孔吸光度(A1)。每孔加入5 μL 酸性溶液。室温摇床孵育20 min。再次用酶标仪测各孔的A值(A2)。计算公式:A3=A2-A1,校正A=各孔A3-空白孔A3。以标准品的校正A作图,浓度值为 X 轴,校正A为 Y 轴,绘制标准曲线。用各样品的校正A在标准曲线上查出相对应的肌酐浓度。
3 统计学处理
所有数据以均数±标准误(mean±SEM)表示。用统计软件SPSS 21.0 进行统计分析。所有实验重复至少 3 次。两组间比较用Student′st检验,多组间比较用单因素方差分析及Bonferroni 或Tamhane′s T2检验。以P<0.05为差异有统计学意义。
结 果
1 CaMKII活性在ADR干预的RTEC中升高
Western blot 结果显示,对比正常培养的RTEC,ADR 干预的 RTEC 中 CaMKII 蛋白磷酸化程度,即CaMKII 活 性(p-CaMKII/t-CaMKII)显 著 增 加(P<0.01),见图1A;同时,在ADR 干预的小鼠肾组织中CaMKII活性也显著增加(P<0.01),见图1B。
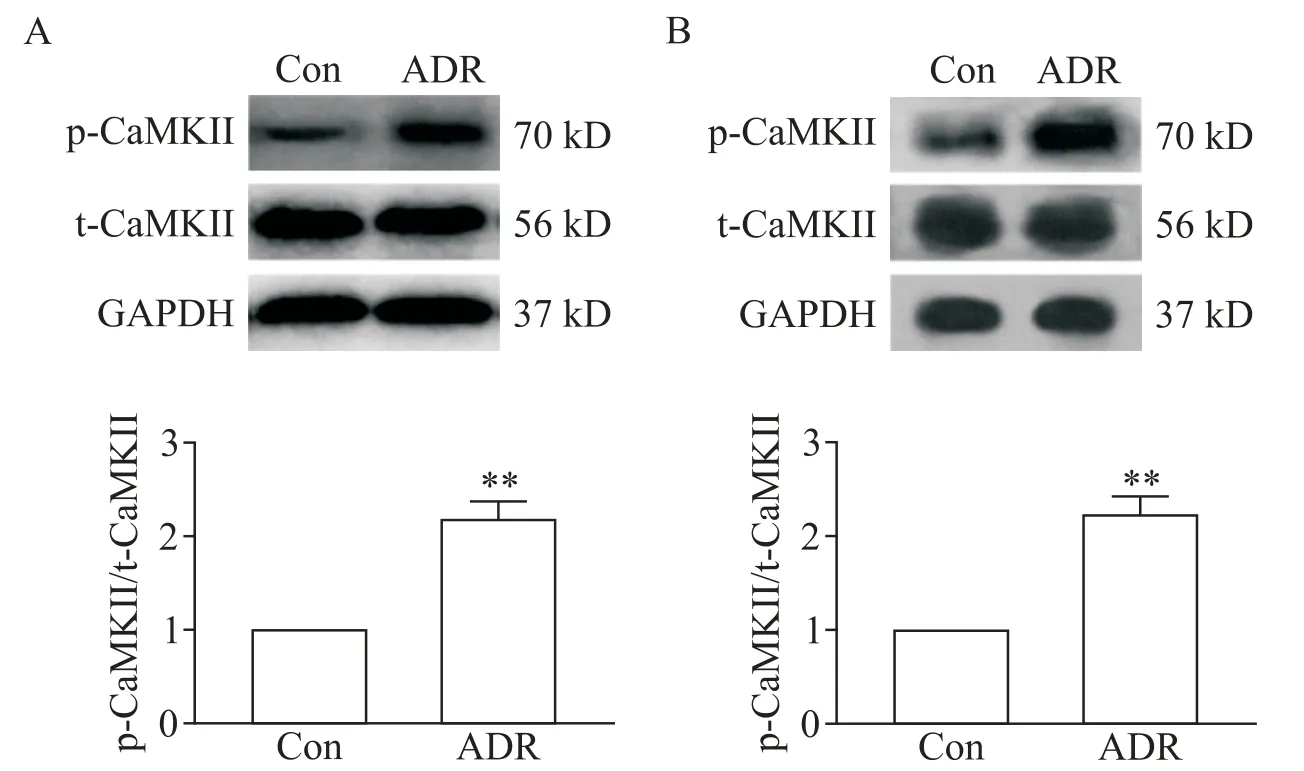
Figure 1. CaMKII activity was increased in ADR-injured RTEC. A:Western blot showed that the CaMKII activity(the ratio of p-CaMKII/t-CaMKII)was increased in ADR-injured HK-2 cells;B:the CaMKII activity was increased in kidney tissues of ADR-treated mice. Mean±SEM. n=5.**P<0.01 vs control(Con)group.图1 CaMKII活性在ADR干预的RTEC中增高
2 抑制CaMKII 活化可减少ADR 诱导的RTEC 凋亡,抑制血清肌酐升高
流式细胞术分析显示,对比Con 组,ADR 组细胞凋亡率显著升高(P<0.01),用K 抑制CaMKII 活化后细胞凋亡率显著降低(P<0.05),见图2A。与此一致地,如图 2B、C 所示,对比 Con 组,ADR 干预的 HK-2细胞和小鼠肾组织中促凋亡蛋白Bax 的表达显著增加,而抗凋亡蛋白Bcl-2 的表达显著减少(P<0.05),抑制CaMKII 活化后此作用显著减弱(P<0.05)。此外,如图2D 所示,对比正常小鼠,ADR 干预小鼠血清肌酐显著升高(P<0.05),予K 后血清肌酐显著下降(P<0.05)。
3 抑制CaMKII 活化可减少ADR 诱导的核NFAT蛋白增加
Western blot 结果显示,对比正常 RTEC,ADR 损伤的HK-2 细胞中,核蛋白NFAT 显著增加(P<0.01),用K 抑制CaMKII 活化后此作用显著减弱(P<0.05),见图3A;同时,对比正常小鼠,ADR 干预小鼠肾组织中核蛋白NFAT 显著增加(P<0.01),予K 后此作用显著减弱(P<0.05),见图3B。
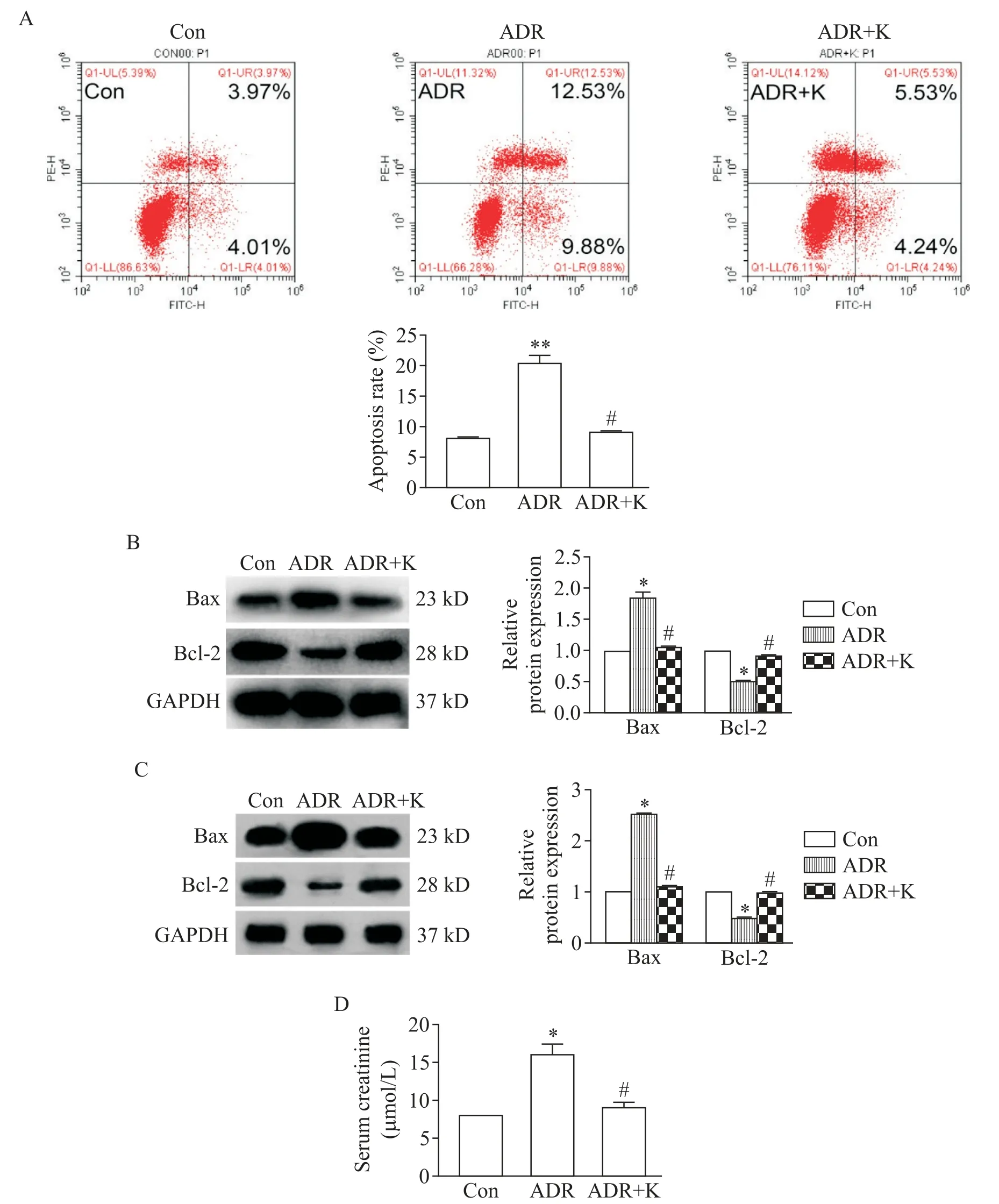
Figure 2. Inhibition of CaMKII activity attenuated ADR-induced RTEC apoptosis. A:cultured HK-2 cells were stained with Annexin V/PI for flow cytometry analysis;B:Western blot showed that Bax protein expression was increased,and Bcl-2 was decreased in ADR-injured HK-2 cells,but inhibition of CaMKII activity by KN-93(K)attenuated these changes;C:Bax protein expression was increased and Bcl-2 was decreased in kidney tissues of ADR-treated mice,but inhibition of CaMKII activity by K attenuated these changes;D:serum creatinine was increased in ADR-treated mice,but it was back to nearly normal after inhibiting CaMKII activity by K in ADR-treated mice. Mean±SEM. n=5.*P<0.05,**P<0.01 vs control(Con)group;#P<0.05 vs ADR group.图2 抑制CaMKII活化可减轻ADR诱导的RTEC凋亡
4 CaMKII可通过NFAT促进RTEC凋亡
流式细胞术结果显示,对比Con 组或siCon 组,ADR 或 CaM 干预 的 RTEC 凋亡率显 著 升高(P<0.05);但在 ADR 或 CaM 干预 RTEC 的同时,加上siNFAT 处理后细胞凋亡率显著降低(P<0.05),见图4。
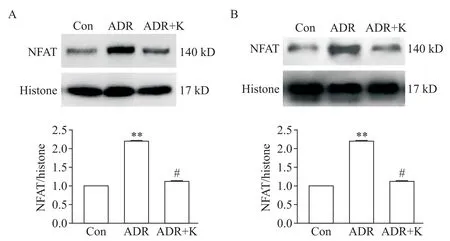
Figure 3. Inhibition of CaMKII activity attenuated ADR-induced increase in nuclear NFAT expression. A:Western blot showed that nuclear protein expression of NFAT was significantly increased in ADR-injured HK-2 cells,but inhibition of CaMKII activity by KN-93(K)attenuated this change;B:nuclear protein expression of NFAT was significantly increased in kidney tissues of ADR-treated mice,but inhibition of CaMKII activity by KN-93(K)attenuated this change. Mean±SEM. n=5.**P<0.01 vs control(Con)group;#P<0.05 vs ADR group.图3 抑制CaMKII活化可减少ADR诱导的核NFAT蛋白增加
讨 论
AKI 严重时可引起严重电解质酸碱紊乱、感染、心衰、肺水肿、昏迷,甚至死亡。AKI 的发病率和死亡率较高[16-17],是危害人类健康的常见急重症之一,在发展中国家造成严重的医疗和经济负担[18-19]。AKI 的早期表现、危险因素和致病机制缺乏认识,常导致AKI 的防治不足,可加速患者病情进展,带来更严重的肾脏损伤、多器官损害和更高的死亡风险[18]。明确AKI 早期发病的分子机制,有助于疾病的早期识别和药物干预。AKI 病因可分为肾前性、肾性和肾后性损伤;其中,肾前性主要是肾灌注减少诱发,肾后性多是尿路梗阻所致;而肾性损伤的病因最为繁多和复杂,包括肾小管、肾间质、肾血管和肾小球的损伤,尤以肾小管的损伤最为多见。RTEC 凋亡是导致肾性AKI 发生发展的重要原因。因此,明确RTEC 凋亡的分子机制有可能为AKI 防治提供参考资料。
丝氨酸/苏氨酸蛋白激酶CaMKII 在各组织广泛表达,与细胞凋亡密切相关[9-10]。药物抑制或基因敲除CaMKII可保护内质网应激诱发的小鼠心肌细胞凋亡[10]。体外研究表明CaMKII 过表达可促进碘海醇诱导的线粒体损伤和RTEC 凋亡[7]。这可能与CaMKII 下游的CypD/mPTP 通路异常激活促进RTEC损伤有关[7]。在上述研究的基础上,为探索AKI 中CaMKII 在RTEC 凋亡中作用和分子机制,我们首先采用ADR 干预体外RTEC和小鼠,结果显示RTEC凋亡率和小鼠血清肌酐显著升高,这与以往研究结论一致[12-13],提示 RTCE 凋亡模型和 AKI 模型构建成功。ADR 干预后HK-2 细胞和小鼠肾组织中CaMKII活性显著增加;抑制CaMKII 活性后,HK-2 细胞凋亡和小鼠肾组织细胞凋亡减轻,小鼠血清肌酐水平回降。上述结果提示CaMKII 异常激活可促进AKI 中RTEC凋亡。
NFAT 是钙调磷酸酶(calcineurin,CaN)的底物,是Ca2+依赖的转录因子。NFAT作为Ca2+/CaM信号通路的下游分子起到促进细胞凋亡的作用[20-21]。高糖刺激下NFAT 核蛋白表达水平显著升高,抑制CaN/NFAT 通路后可显著减轻高糖损伤诱导的肾小球足细胞凋亡[22-24]。此外,Ca2+/CaM 信号下游的 CaMK/CREB 通路也可激活 NFAT 功能[26]。既往研究提示CaMKII 和NFAT 有可能存在相互作用,共同参与调控RTEC凋亡。据此,本研究深入探索上述分子在早期AKI 致病中的作用机制。我们观察到活化剂干预激活CaMKII 后,体外模型中RTEC 细胞凋亡率显著升高,但在沉默NFAT后,细胞凋亡率显著降低。在ADR 损伤的RTEC 和小鼠肾组织中,核蛋白NFAT 显著升高伴随RTEC 凋亡增加,而在抑制CaMKII 活化后核蛋白NFAT 回降且细胞凋亡率降低。上述结果提示CaMKII 可通过调控NFAT 促进小鼠RTEC凋亡。
综上所述,本研究建立了人RTEC 凋亡模型和AKI 小鼠模型,并在模型中观察到CaMKII 可通过NFAT促进小鼠RTEC凋亡。这有可能为AKI防治提供参考资料。

Figure 4. CaMKII promoted RTEC apoptosis through NFAT. A:total NFAT protein expression was knockdown to about 45% in NFAT-siRNA(siNFAT)-treated HK-2 cells;B:apoptosis rate was significantly increased in ADR- or calmodulin(CaM;CaMKII activator)-injured HK-2 cells compared with control(Con)or siRNA blank control(siCon)group,but apoptosis was significantly alleviated by siNFAT in ADR- or CaM-injured HK-2 cells. Mean±SEM. n=4.*P<0.05 vs Con or siCon group;#P<0.05 vs ADR group;△P<0.05 vs CaM group.图4 CaMKII通过NFAT促进RTEC凋亡
猜你喜欢
家庭医药(2018年9期)2018-09-27
中成药(2017年9期)2017-12-19
中成药(2017年5期)2017-06-13
中外医疗(2015年11期)2016-01-04
华南农业大学学报(2015年5期)2015-12-04
哈尔滨医药(2015年5期)2015-12-01
医学研究杂志(2015年8期)2015-06-22
医学研究杂志(2015年12期)2015-06-10
医学研究杂志(2015年4期)2015-06-10
西南军医(2015年6期)2015-01-23
