
HPLC指纹图谱结合模式识别分析评价风湿骨痛胶囊的批间一致性
2021-12-23 11:22陈良妮程雪梅王长虹
中成药 2021年12期
陈良妮,程雪梅,王 琳,高 武,陈 勇,王长虹*
[1.上海中医药大学中药研究所,中药标准化教育部重点实验室,上海市复方中药重点实验室,上海中药标准化研究中心,上海 201203;2.广西中医药大学药学院,广西 南宁 530000;3.国药集团精方(安徽)药业股份有限公司,安徽 宣城 242000]
风湿骨痛胶囊由制川乌、制草乌、甘草、麻黄、红花、木瓜、乌梅制成的中药复方制剂,具有温经散寒、通络止痛的功效,用于寒湿闭阻经络所致的弊病,风湿性关节炎等[1-4]。2015年版《中国药典》对麻黄进行了TLC鉴别、HPLC含量测定;对制川乌、制草乌进行了TLC限量检测及紫外分光光度法测定乌头总生物碱。相关文献利用液质联用技术对方中制川乌、制草乌的9个乌头类生物碱做了定量研究[5]。
指纹图谱是基于对中药及中药制剂物质量群整体作用的认识,通过中药化学成分的光谱或色谱图,实现鉴别中药及中药制剂真实性,评价质量一致性和产品稳定性[6-9]。化学模式识别技术可以对具有模糊性和整体性的中药指纹图谱实现数据降维、识别和分类,是筛选质量差异标志物的重要数学方法,具有较高的预测精度和较强的线性数据分析分类等优势,如主成分分析(principalcomponent analysis,PCA)、偏最小二乘法-判别分析(partial least squares discriminantanalysis,PLS-DA)、判别分析(discriminantanalysis,DA)、偏最小二乘法-判别分析(partial least squares discriminantanalysis,PLS-DA)等,已广泛应用于指纹图谱等多维数据的处理分析中[10-13]。目前,采用指纹图谱结合化学模式识别技术评价不同批次风湿骨痛胶囊的质量稳定性研究还未见报道。本实验通过建立指纹图谱结合化学模式识别技术分析评价风湿骨痛胶囊的质量稳定性和批次一致性。
1 材料
1.1 仪器 Agilent 1260高效液相色谱仪、Agilent G1315C DAD检测器、Agilent Open Lab CDS 2.x化学工作站(美国Agilent公司);AE200电子分析天平(瑞士Mettler-Toledo公司);SK1200H超声波清洗器(上海科导超声仪器有限公司);Milli-Q Intergral水纯化系统(美国Millipore公司)。
1.2 试剂与药物 磷酸、乙腈为色谱纯(美国Fisher公司);水为超纯水;其他试剂均为分析纯。盐酸麻黄碱(批号171241-201809,纯度>98%)、盐酸伪麻黄碱(批号171237-201510,纯度>98%)对照品购于中国食品药品检定研究院;苯甲酰新乌头原碱(批号DST191023-056,纯度>98%)、芹糖甘草苷(批号DST191102-139,纯度>98%)对照品购于乐美天医药/德思特生物公司;绿原酸(批号AF7070309,纯度>98%)、甘草素(批号AF8052202,纯度>98%)对照品购于成都埃法生物科技有限公司;甘草苷(批号070003-201602,纯度>98%)、异甘草苷(批号250026-201511,纯度>98%)、甘草酸单铵(批号070020-201508,纯度>98%)对照品购于上海奈启生物科技有限公司。风湿骨痛胶囊共20批,由国药集团精方(安徽)药业股份有限公司提供,批号分别为180107、180201、180408、180501、180503、180504、180602、181206、190101、190103、190105、190106、190108、190302、190303、190304、190306、190307、190310。
2 方法与结果
2.1 色谱条件 Agilent Zorbax SB-C18色谱柱(250 mm×4.6 mm,5 μm);流动相乙腈(A)-0.2%磷酸(B),梯度洗脱,程序见表1;体积流量1.0 mL/min;柱温30 ℃;检测波长210 nm(0~20 min)、310 nm (20~36 min)、235 nm (36~60 min);进样量10 μL。

表1 梯度洗脱程序
2.2 溶液制备
2.2.1 对照品溶液 称取各对照品适量,70%甲醇溶解,即得(质量浓度分别为盐酸麻黄碱134.6 μg/mL、盐酸伪麻黄碱228.7 μg/mL、绿原酸18.30 μg/mL、苯甲酰新乌头原碱45.30 μg/mL、芹糖甘草苷36.40 μg/mL、甘草苷117.9 μg/mL、异甘草苷31.50 μg/mL、甘草素17.80 μg/mL、甘草酸132.3 μg/mL)。
2.2.2 供试品溶液制备 取装量差异项下胶囊内容物,研细,取约0.8 g,精密称定,置于50 mL具塞锥形瓶中,精密加入70%甲醇10 mL,称定质量,超声(功率250 W、频率40 kHz)处理30 min,放冷,70%甲醇补足减失的质量,摇匀,滤过,取续滤液,即得。
2.3 方法学考察
2.3.1 精密度试验 取同一份供试品溶液,在“2.1”项色谱条件下进样测定6次,以甘草酸峰为参照,测得共有峰相对保留时间、相对峰面积RSD分别在0~0.62%、0.12%~2.85%范围内,表明仪器精密度良好。
2.3.2 重复性试验 取同一批胶囊装量差异项下内容物,研细,平行6份,按“2.2.2”项下方法制备供试品溶液,在“2.1”项色谱条件下进样测定,以甘草酸为参照,测得共有峰保留时间、相对峰面积RSD分别在0.01%~0.33%、0.28%~4.73%范围内,表明该方法重复性良好。
2.3.3 稳定性试验 取同一批胶囊装量差异项下内容物,研细,按“2.2.2”项下方法制备供试品溶液,室温下于0、2、4、8、12、24 h在“2.1”项色谱条件下进样测定,以甘草酸峰为参照,测得共有峰相对保留时间、相对峰面积RSD分别在0.01%~0.23%、0.12%~3.71%范围内,表明溶液在室温下24 h内稳定性良好。
2.3.4 相对保留时间、相对峰面积 20批样品中共有峰相对保留时间RSD在0.01%~0.49%范围内,表明其相对稳定;相对峰面积RSD在3.74%~71.78%范围内,表明其差异较大。
2.4 HPLC指纹图谱建立
2.4.1 图谱生成 取20批样品,按“2.2.2”项下方法制备供试品溶液,在“2.1”项色谱条件下进样测定,将相关数据导入“中药色谱指纹图谱相似度评价系统(2012版)”软件,设置时间窗宽度为0.2 min,采用多点校正法进行全峰匹配,共确定31个共有峰,见图1。

图1 20批样品HPLC指纹图谱
2.4.2 共有峰指认和参照峰选择 吸取“2.2”项下对照品、供试品溶液,在“2.1”项色谱条件下进样测定,结果见图2。通过文献查阅,指认出2号峰为原儿茶酸;通过DAD检测器紫外吸收值比对,指认4号峰为盐酸麻黄碱,5号峰为盐酸伪麻黄碱,8号峰为绿原酸,12号峰为芹糖甘草苷,13号峰为甘草苷,15号峰为苯甲酰新乌头原碱,17号峰为异甘草苷,19号峰为甘草素,22号峰为甘草酸,再选择与相邻色谱峰分离效果良好、保留时间稳定的22号峰(甘草酸)作为参照。
2.4.3 相似度分析 将20批样品相关数据导入“中药色谱指纹图谱相似度评价系统”(2012版)软件,以对照图谱为参照[14],计算相似度,结果见表2。

4.盐酸麻黄碱 5.盐酸伪麻黄碱 8.绿原酸 12.芹糖甘草苷 13.甘草苷 15.苯甲酰新乌头原碱 17.异甘草苷 19.甘草素 22.甘草酸图2 对照品(A)、供试品(B)HPLC色谱图

表2 20批样品相似度
2.5 化学模式识别分析
2.5.1 主成分分析(PCA)将共有峰数据导入SIMICA13.0软件,进行无监督的PCA分析[15],结果见图3。由此可知,20批样品可分为2类,区分度较为明显。
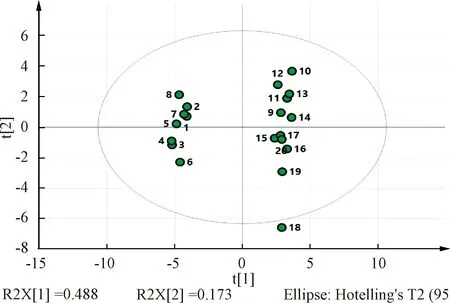
图3 20批样品PCA得分图
2.5.2 偏最小二乘法-判别分析(PLS-DA)将共有峰数据导入SIMCA13.0软件,在PCA分析的基础上建立有监督的PLS-DA判别分析,获得PLS-DA得分图、VIP图、聚类分析图,分别见图4~6。由图4可知,20批样品可分为2个区域。由图5可知,5号峰为伪麻黄碱,19号峰为甘草素,14号峰为麻黄碱,22号峰为甘草酸,15号峰为苯甲酰新乌头原碱,13号峰为甘草苷,2号峰为原儿茶酸,贡献值均大于1;11号峰是制川乌、制草乌、红花、乌梅的共有峰,7号峰是木瓜、乌梅的共有峰,27、30号峰来自甘草,3号峰是制川乌、制草乌共有峰,6号峰来自红花,均对分类有显著影响,即为主要成分的质量标志物。由图6可知,20批样品可聚为2大类,与图4一致。

图4 共有峰PLS-DA得分图

图5 共有峰VIP图

图6 20批样品聚类分析图
3 讨论
本实验对样品处理方法进行考察,确定以70%甲醇超声提取30 min制备供试品溶液。再对检测波长进行考察,发现210 nm(0~20 min)、310 nm(20~36 min)、235 nm(36~60 min)的分段程序检测效果最佳。
20批风湿骨痛胶囊的HPLC指纹图谱中确定31个共有峰,指认出麻黄碱、甘草苷、苯甲酰新乌头原碱等10种成分。采用《中药色谱指纹图谱相似度评价系统》(2012版)软件分析,结果相似度均大于0.95,说明风湿骨痛胶囊质量稳定。但PCA、聚类分析和PLS-DA均将20批样品明显分成2类,结合化学模式识别方法结果,说明不同批次样品共有峰存在一定的差异。调取生产批记录分析发现,20批样品中编号为1~8的采用不同批次的原料药材,而编号为9~20的采用同一批次的原料药材,指纹特征也具有更高的相似性,即该产品的制备工艺具有较高的稳定性,与实验结果完全吻合,说明原料差异对成品的影响是重要因素之一。为了保证生产过程批次间的一致性,应加强对原料药材进行质量控制,并且对质量合格的多个批次原料药材进行校兑后再投入生产也是可供选择的策略之一。
猜你喜欢
基层中医药(2021年3期)2021-11-22
中老年保健(2021年9期)2021-08-24
工程质量(2019年8期)2019-11-15
药学研究(2019年5期)2019-06-05
世界科学技术-中医药现代化(2018年9期)2019-01-29
中国卫生标准管理(2015年4期)2016-01-14
西南医科大学学报(2016年4期)2016-01-03
中国当代医药(2015年33期)2015-03-01
中国药业(2014年20期)2014-05-17
中国刑警学院学报(2014年4期)2014-04-27
