
碳四炔烃选择性加氢催化剂研究进展
2021-12-22 09:13:38孙晋冬展学成吴登峰马好文许昊翔程道建
石油学报(石油加工) 2021年6期
孙晋冬,展学成,吴登峰,谢 元,马好文,许昊翔,程道建
(1.北京化工大学 有机无机复合材料国家重点实验室,北京 100029;2.中国石油 石油化工研究院 兰州化工研究中心,甘肃 兰州 730060)
C4馏分是乙烯裂解装置和炼油裂化装置过程中的副产物,其中C4炔烃(乙烯基乙炔与乙基乙炔)含量较高(质量分数大于35%),易聚合发生爆炸,需要用丁烯、丁烷将C4馏分稀释至安全范围后才能排放火炬或低价出售;此外杂质浓度过高还会严重降低产品质量,造成较大的资源浪费和严重的环境污染[1]。近年来,随着裂解深度及裂解技术等因素的影响,裂解C4馏分中炔烃含量呈增加趋势,导致抽提过程中丁二烯的损失增大、能耗增加。同时随着有机合成工业技术的发展,对丁二烯中炔烃含量的限制更加严格,这些因素均导致丁二烯抽提装置的经济性变差。采用选择加氢除炔技术可将C4炔烃选择加氢转化为丁二烯、丁烯或烷烃,其中1,3-丁二烯是合成橡胶的重要单体,丁烯又是生成甲基叔丁基醚(MTBE)或烷基化的原料,而丁烷又可以返回乙烯装置作为裂解的原料[2]。C4炔烃选择加氢技术不仅可以避免炔烃的排放,大幅降低装置运行的安全风险,还可以回收丁二烯、丁烯或烷烃,提高装置的经济效益。
在C4炔烃选择加氢过程中,催化剂的选择尤为重要,要保证其在一定催化活性的基础上再进一步提高选择性。因此,C4炔烃加氢催化剂的设计与制备一直被广大学者所研究。笔者针对C4炔烃选择加氢催化剂的研究现状及发展趋势进行了综述。
1 混合C4炔烃催化加氢反应
烃类裂解是聚合用烯烃的主要来源,裂解过程产生的炔烃杂质是烯烃聚合反应的毒物,在聚合反应前必须深度脱除,聚合烯烃一般要求炔烃质量分数低于0.001%,否则会引发副反应,降低产品质量和收率[3]。通过催化加氢的方式从混合C4中去除炔烃被认为是一种比较高效的方法,也是去除聚合原料中炔烃的关键反应之一。在混合C4的催化加氢反应中,采用经典的Horiuti-Polanyi机理可以得到丁烯和丁烷等多种产物[4]。如图1(a)所示,部分乙烯基乙炔加氢转化为二烯烃及乙基乙炔加氢形成丁烯是去除混合C4资源中炔烃,使之变废为宝的重要途径。其中,1,3-丁二烯作为一种简单的不饱和共轭烃,又被广泛用作机理研究的探针分子。
根据Horiuti-Polanyi提出的反应机理,以1,3-丁二烯为例分析其选择加氢反应机理。图1(b)显示了1,3-丁二烯选择加氢为丁烯可能主要涉及的两种中间体:一种是H加入到内部原子上会形成一个1-丁烯-4自由基(1B4R);另一种是H加入到末端碳原子上会形成2-丁烯-1自由基(2B1R)[5]。通过鉴定不同中间体的存在,可以有效辨别丁烯形成的反应途径。
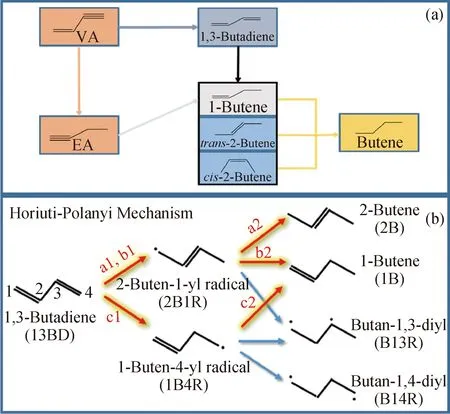
VA—Vinyl acetylene;EA—Ethyl acetylene图1 混合C4炔烃加氢反应路径及反应机理[4-5]Fig.1 Hydrogenation reaction path and reaction mechanism of mixed C4 alkynes[4-5](a)The basic reaction path of mixed C4 selective hydrogenation;(b)Schematic diagram of the Horiuti-Polanyi mechanism of partial hydrogenation of 1,3-butadiene
2 C4馏分选择性加氢催化剂
目前,C4馏分选择性加氢催化剂主要分为贵金属催化剂与非贵金属催化剂两大类。其中,贵金属催化剂主要以Pd系催化剂为主,而非贵金属催化剂主要以Cu系与Ni系催化剂为主。
贵金属Pd因其良好的吸氢储氢能力,在C4馏分选择性加氢工艺中得以广泛应用[6]。与Pd催化剂不同,非贵金属Cu基催化剂在C4炔烃加氢反应中表现出较高的选择性[7],但Cu基催化剂较短的使用寿命和较低的催化活性难以满足工业要求。而非贵金属Ni系催化剂最大的优势是价格低廉,但其对炔烃的选择性较差,因此Ni一般作为一种助剂来调节催化剂的性能[8]。与非贵金属催化剂相比,Pd系催化剂的工艺操作条件更为温和,并且表现出更好的炔烃催化加氢活性。采取合理的策略对其进行改性,还可以提高其选择性和抗毒性。因此Pd系催化剂最适合作为C4炔烃选择加氢催化剂。
炔烃的选择性加氢是制备烯烃的一种有效方法,在C4资源的充分利用领域具有特殊的意义[9]。在丁二烯加氢制丁烯的反应中,Pd催化剂因具有高效的加氢催化效果而成为催化领域的研究热点。目前,学术界广泛的共识就是需要对Pd系催化剂进行改性来提高其作为炔烃选择性加氢的催化性能[10]。Pd催化剂虽然在C4馏分加氢反应中表现出较高的活性,但是其选择性较低,这会导致C4炔烃过度加氢生成大量的副产物,最终影响加氢产品的质量,从而限制了其在工业生产中的大规模应用。近几年来,学术界研究发现,对Pd催化剂进行适当的改性,可以提升其对C4馏分中炔烃组分的加氢选择性,满足C4炔烃选择性加氢的产品指标[11]。通过采取在Pd催化剂体系中引入助剂、对载体改性以及对催化剂活性组分进行尺寸与形貌调控等策略,来改善Pd组分与炔烃之间的相互作用,可提高其对目标产物的选择性。
将Ni、Cu、Pb等助剂引入Pd催化剂体系是一种行之有效的优化催化剂性能的方式[12],第二金属组分的引入改变了活性组分Pd的几何结构和电子结构,使Pd晶体表面对反应物的吸附能力发生变化,从而改善了Pd催化剂的活性与选择性。其次,在制备氧化铝载体过程中改变载体的比表面积、孔体积、孔径及酸性中心也能改善催化剂的性能[13]。在C4馏分选择性加氢反应中,载体比表面积在110 m2/g 左右时,催化活性与选择性较好。载体比表面积大,能够提供的活性中心多,催化剂的活性高,但选择性差,二烯烃损失严重,且载体的酸性中心又会催化炔烃和烯烃发生聚合反应生成绿油,堵塞孔道,造成催化剂的失活[14]。因此载体适宜的比表面积和低酸性中心利于延长催化剂的使用寿命以及提高催化剂的活性与选择性。另外,通过对Pd纳米粒子(Pd-NPs)的尺寸与形貌进行调控,在催化剂的制备过程中添加封端剂、稳定剂与还原剂,并严格控制各物质的摩尔比、反应液的温度与pH值等条件,可以稳定制备出不同尺寸与形貌的Pd-NPs[15]。C4炔烃选择性加氢反应是结构敏感反应,Pd纳米粒子的粒径并非是越小越好,因此将纳米粒子的尺寸调控到一定的范围可以获得更好的催化效果。研究表明,在C4炔烃选择性加氢反应中,纳米粒子的粒径在4~10 nm之间时,催化效果较好[16]。另外,研究发现Pd/Al2O3催化剂的活性和选择性与Pd(111)晶面活性位点的总量有关[17],通过控制合成具有特定晶面暴露的Pd催化剂可有效调控丁二烯加氢反应的活性和选择性。
3 C4炔烃加氢工业催化剂优化策略
近年来,C4炔烃选择加氢催化剂的主要研究方向逐渐由负载型金属纳米催化剂向单原子催化剂转变。单原子催化剂较负载型纳米粒子催化剂有更好的活性和选择性,因此单原子催化剂在近几年的发展特别迅速。
3.1 负载型金属纳米粒子催化剂
3.1.1 载体预处理
负载型金属纳米粒子催化剂的载体主要是氧化铝、二氧化硅、二氧化钛、碳纳米管或分子筛等。与其他载体相比,Al2O3因价格廉价、性能稳定而被广泛应用[18]。但是,Al2O3的表面存在酸性中心,易引发不饱和烃的聚合反应,导致催化剂孔道堵塞,造成催化剂失活。另外,不同类型氧化铝的表面酸性中心也不同,γ-Al2O3较α-Al2O3拥有更大的比表面积,但同时其也拥有更强的酸性中心。此外,煅烧温度也会对氧化铝结构性质产生影响,不同煅烧温度下制备的Al2O3载体具有不同的比表面积和酸性中心[19]。因此现有研究侧重于调节载体的表面酸性来提高催化剂性能。另外,研究表明,通过预处理手段得到的复合载体具有更加优越的性能,如Al2O3-TiO2不仅具有更大的孔径,利于催化剂活性位点与反应物的接触;而且还具有更大的比表面积,提高了催化剂的抗中毒能力[20-21]。
3.1.2 助剂改性
随着纳米科学的发展,负载型金属纳米催化剂在工业生产中得以广泛应用。C4炔烃选择性加氢反应常常伴随着过度加氢生成饱和烃的副反应,部分催化剂的化学选择性并不理想。如果将第二金属(如Ag、Cu、Ni)引入到纳米催化剂体系中,纳米催化剂的化学选择性可大大提高,添加助剂改性成为改进催化剂综合性能,特别是提高选择性的有效手段。目前研究甚为广泛的改性助剂为Ag、Cu,Ag的加入可以有效提高催化剂的寿命,但也降低了其催化活性;而Cu助剂的加入则需要较高的反应温度,但高温容易导致原料严重结焦,降低了催化剂的稳定性。
贵金属催化剂在催化选择性加氢反应中,由于其较强的氢活化作用,通常对烯烃的选择性较低。为了提高烯烃的选择性,目前应用最广泛的方法之一是通过引入第二种非贵金属助剂,形成贵/非贵金属双金属催化剂,如Pd-Ni、Pd-Cu、Pd-Co、Pd-Zn、Pt-Cu等。在已有的研究中,合成的负载在氧化铝上的Pt-Cu合金催化剂,在丁二烯选择性加氢反应中,在全转化温度下(T100%)表现出99%的丁烯选择性[22]。
(1)钯铜合金催化剂
在Pd(111)、Cu(111)和Pd1Cu3(111)晶面进行密度泛函理论计算,以了解催化剂的表面性质并比较1,3-丁二烯及其氢化产物和氢气的吸附能[23],结果见表1。由表1可知,H在Pd1Cu3(111)晶面的吸附强度最弱,而不同C4分子在Pd1Cu3(111)晶面的吸附强度则居于Pd(111)和Cu(111)之间,说明Cu组分的引入实现了反应物和产物分子在催化剂表面的吸附强弱调控,进而可能影响反应路径。同时,由于单烯在各晶面上的吸附强度明显弱于丁二烯,说明单烯烃产物形成后比反应物更容易从催化剂表面脱附。Insorn等[24]在Pd/Al2O3催化剂中加入了适量的Cu改善了催化剂对乙烯基乙炔加氢反应的催化性能。该研究表明,在制备的Pd-Cu/Al2O3催化剂中,当Pd和Cu摩尔比为3∶2时,Pd-Cu/Al2O3催化剂催化速率最高,说明Cu的引入提高了催化剂的催化活性。此外,在反应过程中,Cu的存在也延缓了催化剂上的炭沉积,大大延长了催化剂的使用寿命。

表1 通过DFT计算得出H和不同C4分子在Pd(111)、Cu(111)和Pd1Cu3(111)晶面上的最低吸附能[23]Table 1 Minimum adsorption energy of H and different C4 molecules on Pd(111),Cu(111)and Pd1Cu3(111)crystal planes calculated by DFT[23] Ea/eV
(2)钯银合金催化剂
关于合金化PdAg纳米催化剂的合成这一类的报道并不多见,由于试剂选择、制备条件的限制等因素,制备的催化剂总是存在尺寸形貌不可控、分散度较差以及合金化程度低等问题,因此大部分的PdAg合金催化剂在实际生产中并不实用。Troutman等[25]报道的多元醇辅助法,以乙二醇为溶剂,聚乙烯吡咯烷酮(PVP)为保护剂,在150 ℃的条件下成功合成尺寸范围为3.6~6.0 nm,并且活性组分显示出高单分散性的PdAg合金纳米催化剂,如图2所示。Ma等[5]在1,3-丁二烯加氢反应中,在Pd表面引入Ag可以减弱1,3-丁二烯和1-丁烯在PdAg催化剂表面的吸附,从而提升1-丁烯的选择性。在PdAg合金催化剂中Ag含量的增加可以提高1,3-丁二烯加氢的催化性能,这为石油炼制中1,3-丁二烯选择性加氢的PdAg合金纳米催化剂的设计提供坚实的基础。
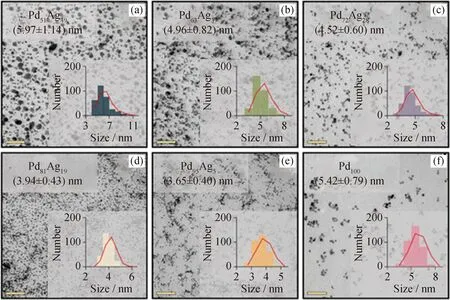
图2 PdxAg100-x纳米合金催化剂的透射电镜图像及其粒径分布[25]Fig.2 TEM images and particle size distribution of PdxAg100-x nano-alloy catalysts[25](a)Pd51Ag49,particle size:(5.97±1.14)nm;(b)Pd63Ag37,particle size:(4.96±0.82)nm;(c)Pd72Ag28,particle size:(4.52±0.60)nm;(d)Pd81Ag19,particle size:(3.94±0.43)nm;(e)Pd95Ag5,particle size:(3.65±0.40)nm;(f)Pd100,particle size:(5.42±0.79)nm
(3)钯镍合金催化剂
不同Pd和Ni摩尔比的PdxNiy/ZnO催化剂的XRD谱图及加氢性能如图3所示。由图3(a)可知,在42.1°、41.6°和41.0°处的衍射峰分别对应Ni和Pd摩尔比为3∶1、1∶1和1∶3的PdxNiy合金的(111)晶面衍射峰。与单金属Pd催化剂相比,随着Ni含量的增加,PdNi双金属催化剂的衍射峰向右发生了偏移,表明了PdNi合金结构的形成[26]。由图3(b)可知,在1,3-丁二烯选择性加氢性能评价中发现,相较于单金属Pd、Ni催化剂,使用双金属PdNi/ZnO催化剂,丁烯的选择性和转化率都有显著提高。双金属Pd1Ni1/ZnO催化剂表现出优异的加氢性能,其中丁烯总选择性为88.93%;然而单金属Pd/ZnO催化剂的丁烯总选择性仅为46.92%,这就是双金属协同作用的结果。因此,Pd1Ni1/ZnO催化剂的合金化可以有效防止丁烯进一步氢化,增加对丁烯的选择性。

图3 不同Pd和Ni摩尔比的PdxNiy/ZnO催化剂的XRD谱图及加氢性能[26]Fig.3 XRD patterns and hydrogenation performance of PdxNiy/ZnO catalysts with different Pd/Ni molar ratios[26](a)XRD diagram;(b)Hydrogenation performance
(4)混合C4加氢反应中的Pd基合金双金属催化剂
图4为混合C4加氢中的Pd基合金双金属催化剂的加氢性能。由图4可知,在乙烯基乙炔混合C4液相加氢反应中,向Pd催化剂中加入适量的Ag、Ni和Mn可以改变Pd催化剂的催化性能[27]。根据1,3-丁二烯的选择性和收率,催化剂催化性能由大到小的排序为PdMn/Al2O3、Pd/Al2O3、PdNi/Al2O3、PdAg/Al2O3。在Pd催化剂中加入Ni只能提高活性;而加入Ag甚至会抑制活性;加入Mn则显著提升了催化性能。特别的,当Pd/Mn摩尔比为2∶1时,PdMn催化剂的催化活性大幅提升,并适度提高了1,3-丁二烯的收率和选择性。
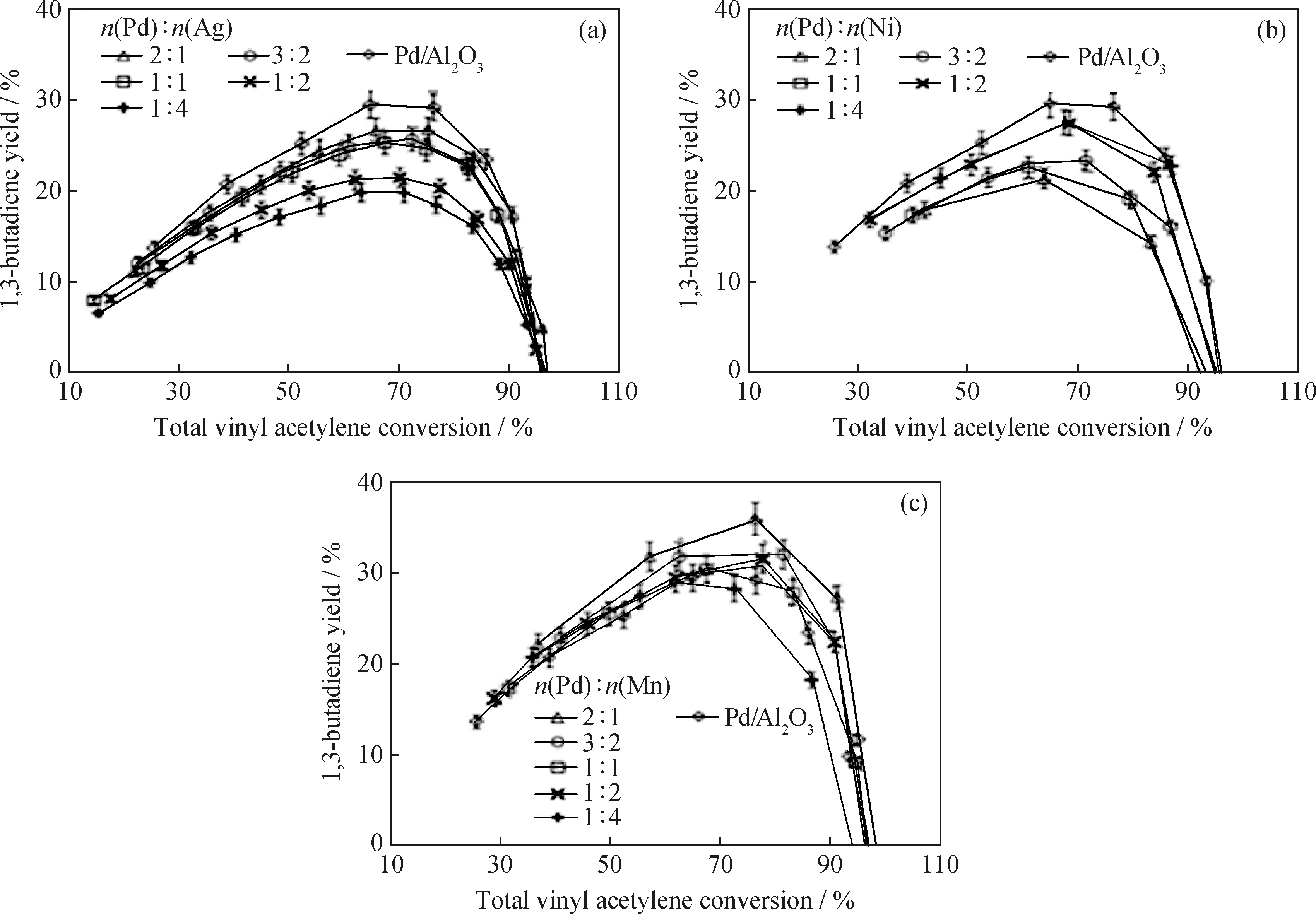
图4 混合C4加氢反应中的Pd基合金双金属催化剂的加氢性能[27]Fig.4 hydrogenation performance of Pd-based alloy bimetallic catalyst in mixed C4 hydrogenation process[27](a)PdAg/Al2O3;(b)PdNi/Al2O3;(c)PdMn/Al2O3
3.1.3 微观调控
研究表明,负载型M-NPs催化剂的尺寸和形貌是影响其催化性能的关键因素[28]。而利用多元醇法或胶体法可以实现金属纳米粒子尺寸与形貌的可控制备[29]。一般来说,贵金属的微观结构和其催化性能息息相关,特别是在催化反应中[30]。金属纳米粒子的小晶面与封端剂的协同效应是产生不同形貌纳米粒子的重要因素。而金属纳米粒子对C4加氢有着很大的尺寸效应与晶面效应,结构敏感反应以及晶面的吸附能力差异都决定着其催化性能。因此可以通过优化金属纳米颗粒的尺寸与形貌使金属纳米颗粒的催化性能更上一层楼。
(1)尺寸调控
图5中给出了Pd纳米胶体与对应的负载型催化剂的TEM图以及每个样品的Pd颗粒尺寸分布图。由图5可以明确地看出,Pd胶体的颗粒粒径要小于负载型Pd催化剂的粒径。这主要是由于负载型催化剂在制备过程中需要经过高温焙烧的手段来增强金属活性组分与载体之间的相互作用,提升催化剂的性能。而在高温焙烧的过程中,又会使得Pd纳米颗粒聚集生长[31]。图5(a)中PVP/Pd(PVP为聚乙烯吡咯烷酮)摩尔比为20,图5(c)中PVP/Pd摩尔比为80,比较两图可以得出,PVP与Pd的摩尔比会严重影响Pd-NPs的尺寸。图5(c)显示的Pd-NPs的尺寸明显小于图5(a)中的尺寸,且粒径分布也更为均匀。这是由于PVP作为金属纳米粒子的保护性聚合物,通过C=O基团与Pd原子的配位作用,大量的PVP吸附在Pd2+的表面,从而阻止Pd粒子聚集以形成更加细小的Pd-NPs[32]。

图5 Pd催化剂的TEM图和相应的尺寸分布图[31]Fig.5 TEM images of Pd catalyst and corresponding size distribution diagram[31](a),(c)Pd colloidal catalyst;(b),(d)Pd supported catalyst
(2)形貌调控
在C4炔烃加氢反应中,尽管载体改性、添加助剂仍是提升催化剂加氢性能的重要手段,但是随着胶体技术的发展,形貌可控的纳米粒子催化剂也逐渐显现出了它独特的加氢性能。通过控制催化剂粒子的形貌来调节活性位点的结构,更有助于提升催化剂的活性和选择性。Martins等[33]通过胶体合成制备出尺寸为11 nm的均匀分布的Pd纳米立方体(如图6(b)),其暴露的晶面主要由(100)面组成。利用此方法制备的Pd/SO2催化剂在1,3-丁二烯加氢中表现出不错的加氢性能。在1,3-丁二烯初始转化率相同的情况下,Pd纳米立方体催化剂对丁烯的选择性高达100%,而传统浸渍法制备的Pd催化剂对丁烯的选择性仅为70%左右;随着丁二烯转化率的提高,相较于传统浸渍法制备的Pd催化剂而言,有无载体的Pd纳米立方体催化剂的活性与选择性都明显更高(如图6(e))。这可能是由于传统浸渍法制备的催化剂的金属表面表现出更不均匀的晶体结构,而纳米立方体主要表现出(100)暴露面所致。此外,Piccolo等[34]利用胶体法成功制备出由(111)面组成的Pd纳米八面体。在1,3-丁二烯加氢反应中,主要暴露(111)面的八面体比主要暴露(100)面的纳米立方体表现出更好的催化性能。通过对1,3-丁二烯及其产物的吸附能分析发现,八面体比立方体有着更低的吸附能,这就意味着单烯生成后更容易脱附,避免过度加氢形成烷烃。
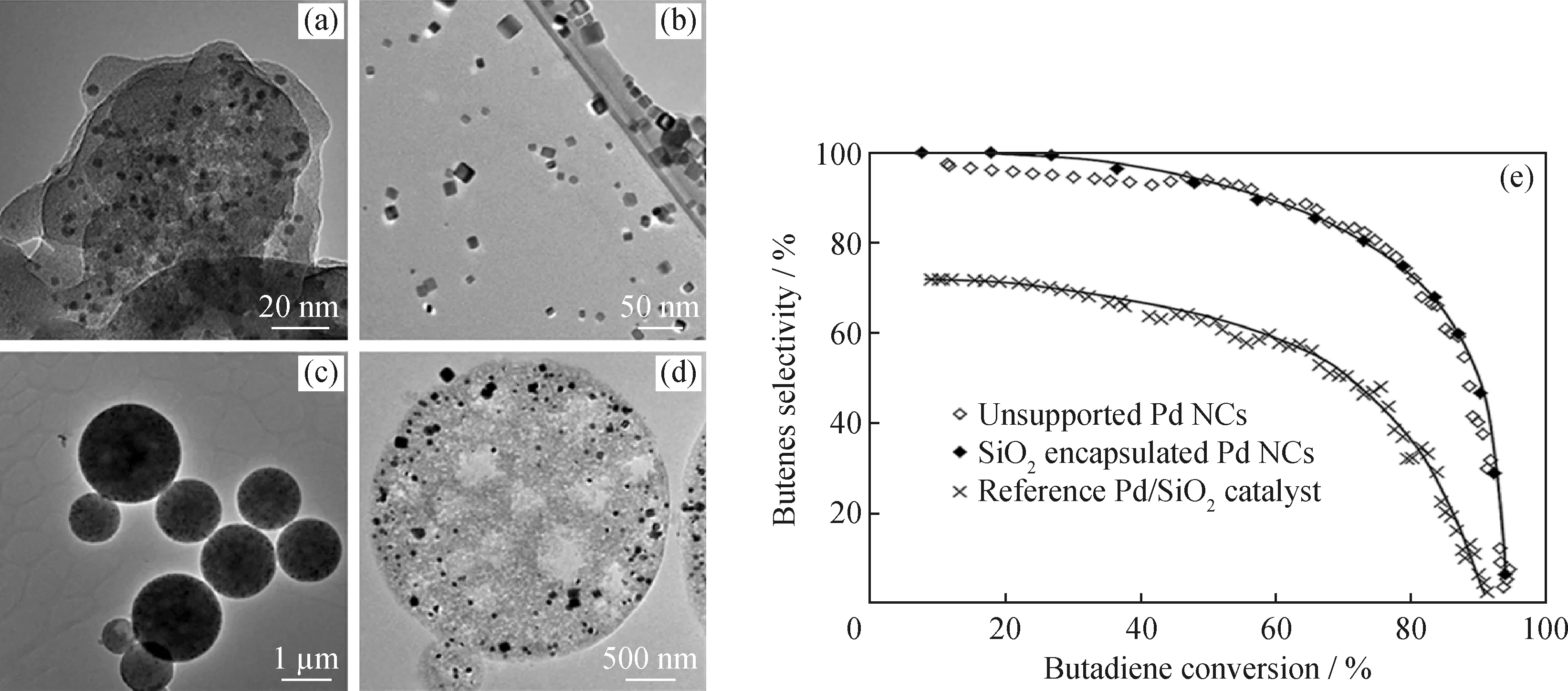
图6 Pd纳米立方体的TEM照片及其各催化剂对1,3-丁二烯的加氢性能[33]Fig.6 TEM images of Pd nanocube and hydrogenation performance of each Pd catalyst on 1,3-butadiene[33](a)Reference Pd/SiO2 impregnated sample;(b)Unsupported Pd nanocubes;(c),(d)SiO2-encapsulated Pd nanocubes;(e)Hydrogenation performance
目前,多元醇法与胶体法是对金属纳米粒子尺寸与形貌调控的最为有效的方法。Favier等[35]在多元醇介质中成功控制合成Pd纳米颗粒,并重点介绍了其在尺寸、形貌和表面状态等方面的研究进展。由于PVP在(111)和(100)面上的选择性吸附,在乙二醇中可以形成不同形貌的Pd-NPs结构;并且在溶液中控制Pd/PVP的摩尔比,制备出了尺寸在3.5~5.5 nm的球形纳米粒子。Wu等[36]采用多元醇法对金属Pd-NPs进行形貌调控,对温度、前驱体浓度、PVP浓度和乙二醇与水的体积比等条件进行严格控制,成功合成了均匀性较好的10 nm的Pd纳米立方体以及长径比一定的Pd纳米棒。Hu等[37]在乙醇与水的溶剂中,成功制备了3.8~7.6 nm的Pd-NPs。由于乙醇的还原速率较慢,硼氢化钠作为强还原剂,两者共同作用下,Pd纳米粒子的粒度细小且均匀。将所制备的Pd纳米催化剂用于C4炔烃选择性加氢反应中,加氢活性以及选择性都有所提升。鉴于优化催化工艺设计需要同时考虑不同层次的要求,不同尺度(纳米、微观和宏观)与不同形貌的纳米颗粒催化剂在C≡C到C=C加氢反应中的应用效果突出[38],并受到越来越广泛的关注。
3.2 单原子催化剂
随着高分辨电子显微镜技术以及表征技术的发展,人们得以观察到纳米、亚纳米甚至原子级的物质。通过观察,已经可以推断出,当金属粒子的尺寸缩小到1 nm以下时,其电子性质会发生巨大的变化(如图(7))[39]。在金属纳米催化剂中,如果纳米颗粒的尺寸缩小到原子尺度,每一个金属原子直接与载体中的杂原子相互作用,提高了催化剂暴露的活性位点数量,那么其催化活性必能得到大幅度提升。就加氢反应而言,亚纳米、原子金属粒子与反应物的相互作用较纳米粒子会出现很大的差异,从而表现出不同的催化性能。单原子催化剂(SACs)作为一种特殊的金属负载型催化剂,催化剂上的所有金属活性组分都是以原子的形式孤立地分散在载体上,没有形成任何的团簇或纳米粒子[40]。2011年,Qiao等[41]首次提出了单原子催化的概念,他们制备的单原子Pt1/FeOx催化剂在CO氧化和CO选择性氧化反应中表现出很高的催化活性和稳定性。随后,研究学者发现金属纳米粒子的尺寸是影响催化剂活性的关键因素[42]。在此基础上,开发了用于选择性氢化反应的新一代催化剂:单原子催化剂。因为活性金属的最终分散性和活性物种的均匀组成,Pd-SACs在苯乙烯加氢[43]和乙炔选择性加氢制乙烯[44]等加氢反应中表现出优异的催化活性和选择性。
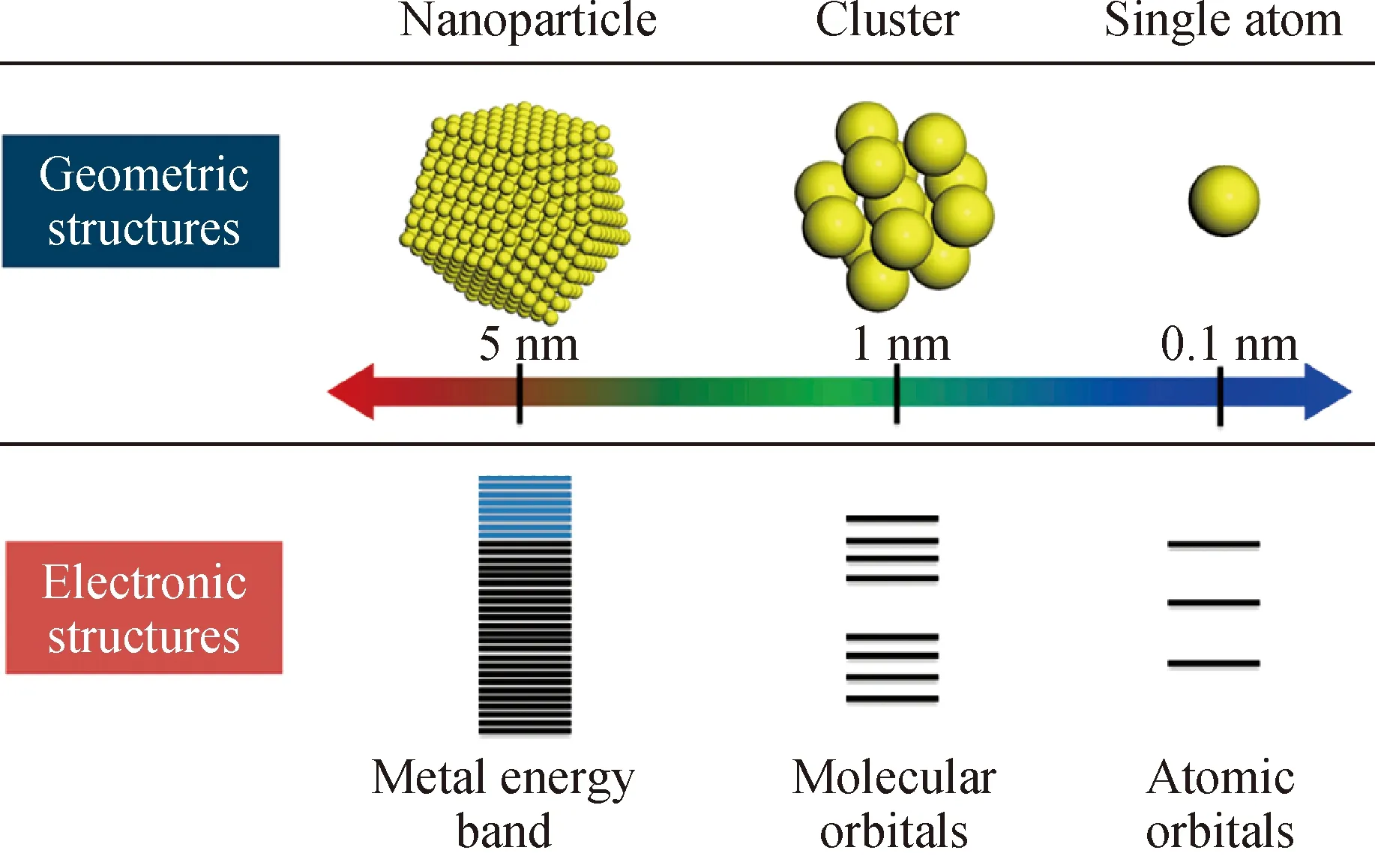
图7 单原子、团簇和纳米粒子的几何和电子结构[39]Fig.7 Geometric and electronic structures of single atoms, clusters and nanoparticles[39]
在单原子Pd1/石墨烯催化剂与3种Pd纳米粒子催化剂进行1,3-丁二烯加氢性能对比中发现,丁烯选择性是丁二烯转化率的函数;并且单原子Pd催化剂相较于其他3种Pd纳米粒子催化剂而言,在保持丁二烯100%转化率的前提下仍能获得80%以上的丁烯选择性(图8(a))。在丙烯存在下,4种催化剂对丙烯的转化率以及在1,3-丁二烯转化率为98%时丁烯的分布如图8(c)、8(d)所示。单原子Pd催化剂对丙烯的转化率最低,仅为0.1%;并且在1,3-丁二烯98%转化率的前提下对丁烯选择性100%,尤其是对1-丁烯选择性高达69%。由此推断出丁二烯是单π吸附模式,因为单π吸附模式不利于同时对丁二烯的2个双键加氢,难以形成丁烷,从而保障了对丁烯的高选择性(如图6(e))[39]。通过DFT计算可知,1,3-丁二烯的吸附作用(-0.98 eV)明显强于丁烯(-0.86、-0.82、-0.76 eV),这就使得在加氢反应期间,1,3-丁二烯在Pd原子上占据明显的优势(图8(f))。此外,几何效应导致丁二烯在Pd原子上的堆积密度高于Pd纳米粒子上的堆积密度,这将进一步增强空间效应,从而更有效地抑制二次氢化反应。Yan等[45]运用原子层沉积的方法(ALD)成功制备了石墨烯负载的Pd-SACs,并将其运用于1,3-丁二烯选择加氢实验。实验结果表明,Pd-SACs 不仅加氢活性高,而且对1-丁烯的选择性高达71%。通过探究其加氢机理得知,当Pd颗粒尺寸减小到单个原子尺度时,会显著增加丁二烯对H2的强烈吸附所引起的空间位阻效应。由于空间位阻的增强会弱化1,3-丁二烯分子在Pd单原子上的吸附,可以更有效地减弱丁烯在二次加氢过程中的吸附。

图8 单原子Pd催化剂的选择性加氢性能及在烯烃上吸附能的DFT计算[39,45]Fig.8 Selective hydrogenation performance of monatomic Pd catalyst and DFT calculation of adsorption energy on olefins[39,45](a)-(d)show the catalytic performance of Pd1/graphene,Pd-NPs/graphene,and Pd-NPs/graphene at 500 ℃ and Pd/carbon catalysts for the selective hydrogenation of 1,3-butadiene;(e)shows the hydrogenation mechanism of 1,3-butadiene over Pd single-atom catalyst;(f)shows the adsorption configuration of H2,1,3-butadiene,1-butene,cis-2-butene,and trans-2-butene on Pd1-O/graphene and their corresponding adsorption energies
Li等[46]报道了热解配位聚合物(PCP)策略制备的一系列单原子位点(SAS)催化剂,包括SAS-Fe、SAS-Ni、SAS-Cu、SAS-Zn、SAS-Pd和SAS-Pt等。对于C4炔烃选择加氢催化剂的应用主要以Ni、Pd催化剂为主,尽管Ni、Pd单原子催化剂的实验室研究已经取得了不错的进展,但是在走向工业化的道路上仍存在一定问题。此前文献中报道的单原子催化剂都存在金属负载量低、制备过程复杂且可重复性较低等问题。随着PCP策略的出现,实现了单原子催化剂的克级生产,这无疑是单原子催化剂在选择加氢反应中早日实现工业应用的巨大突破。
4 结语与展望
尽管先前的文献中已经报道了多种提升Pd基催化剂催化性能的方法,但由于制备技术的不成熟,在实际工业应用中主要还是停留在载体与助剂的改性上。首先,载体与助剂的改性虽然在一定程度上提升了催化剂的性能,但这也会让制备过程更加复杂化且甚至需要引入有毒或价格昂贵的前体。Büchele等[47]表明使用有缺陷的多孔氮化硼载体可以有效控制Pd纳米粒子,使之呈现高分散状态,并且在炔烃的选择性加氢中产生良好的催化性能。此外,第二金属的加入与Pd发生合金化效应,改变了Pd的电子结构,使催化性能得到提升。
第二,采用聚乙烯吡咯烷酮作为稳定剂的胶体法已被证明适用于制备尺寸与形貌可控的Pd-NPs。为了防止纳米粒子的团聚需要加入表面活性剂(PVP,聚乙烯吡咯烷酮),而表面活性剂会堵塞催化剂的活性位点,在不改变纳米粒子的尺寸、形貌和稳定性的情况下去除表面活性剂就显得尤为重要。目前,去除PVP的方法很少,如热氧化法[48]和紫外臭氧法[49]。对于热氧化处理,需要较高的温度,这会严重影响纳米颗粒的尺寸和形貌,导致催化性能的大幅变化。在紫外臭氧处理中,Pd-NPs表面PVP的去除过程往往难以控制。Yang等[50]提出的蒸汽处理法,不仅能有效地去除Pd-NPs表面的PVP,而且能很好地保持Pd-NPs的原有结构,显著地提高其催化性能。该方法有望促进胶体法制备的钯纳米粒子催化剂在加氢领域的广泛应用。
第三,单原子催化剂(SACs)的活性位点孤立、金属负载量低等问题限制了它的发展,仍需要继续优化合成方法来达到SACs的大规模稳定制备的目的。Gan等[51]报道了一种简便、经济、可扩展的球磨方法来制备千克级氧化物负载的SACs,成功合成大批量(>1 kg)Au1/CeO2-SACs。此外,该方法也可以扩展到制备一系列氧化物负载的贵金属SACs。He等[52]提出了一种机械化学方法来制备贵金属SACs,此方法不需要模板、添加剂或溶剂,这大大降低了制造工艺的复杂性,从而降低了成本。这些SACs的合成策略为负载型贵金属单原子催化剂的发展指明了方向。
目前国内C4炔烃的综合利用效率不高,存在大量的资源浪费与环境污染问题,因此选择性加氢催化剂的设计与制备依旧是目前工业催化研究的热点与重点。然而由于催化剂加氢活性与选择性的问题,选择加氢催化剂的设计与制备仍是一个充满挑战的工作。近几年来基于广大学者的研究,C4炔烃选择加氢催化剂的性能得到了一定的改善。从金属纳米粒子催化剂到单原子催化剂,每一次的改进都伴随着催化性能的改善。充分了解加氢反应机理后,若能实现纳米尺度的形貌调控以及原子尺度的SACs催化剂的可控制备,Pd系催化剂在C4炔烃加氢反应体系的催化性能必定能够得到巨大的提升。
猜你喜欢
上海师范大学学报·自然科学版(2023年1期)2023-06-30 07:18:19
中国特种设备安全(2022年1期)2022-04-26 14:16:10
石油炼制与化工(2017年2期)2017-04-07 08:40:11
石油炼制与化工(2017年1期)2017-04-06 07:18:53
化工管理(2017年18期)2017-03-03 16:40:34
化工设计通讯(2017年9期)2017-03-02 16:22:38
赣南师范大学学报(2016年3期)2016-07-18 05:51:05
中国塑料(2016年6期)2016-06-27 06:34:04
物理化学学报(2015年5期)2015-02-28 17:34:59
化工装备技术(2014年3期)2014-04-08 13:18:08
