
UPLC-MS/MS测定水和土壤中唑啉草酯及其代谢物的方法验证
2021-12-22 01:51杨志富欧晓明曾利红侯根连
现代农药 2021年6期
杨志富,欧晓明,曾利红,汤 涛,梁 骥,侯根连,欧 将
(1.湖南化工研究院有限公司国家农药创制工程技术研究中心,长沙 410014;2.农用化学品湖南省重点实验室,长沙 410014;3.湖南化研院检测技术有限公司,长沙 410014;4.浙江省农业科学院农产品质量标准研究所,杭州310021)
唑啉草酯属于先正达开发的一种新苯基吡唑啉类除草剂,作用机理为通过抑制乙酰辅酶A羧化酶,造成植物脂肪酸生物合成受阻,使细胞生长分裂停止,破坏细胞膜含脂结构,导致杂草死亡。我国已于2010年登记用于防除冬小麦和春小麦以及大麦田一年生禾本科杂草[1-3]。唑啉草酯应用于田间后,主要的代谢产物有M2和M3,母体和代谢物均可能随雨水、淋溶等进入自然水体和土壤中,影响生物生长从而造成生态风险[4-5]。建立唑啉草酯及其代谢物M2、M3在水和土壤中的分析检测方法,旨在为该药剂在环境中的研究及风险评估提供技术支持。
目前关于唑啉草酯的报道多为合成[6-7]、应用技术[8-10]、质量控制[11]和在植物基质中的残留分析[12],未见同时对其母体和代谢物在水和土壤中的检测方法。农药在环境介质中的残留量一般较低,普通的液相色谱分析方法的检出限难满足分析要求。笔者建立的超高效液相色谱-串联质谱分析方法具有更高的灵敏度,经验证可用于唑啉草酯及其代谢物M2、M3的环境归趋和环境毒理试验中农药残留分析。
1 材料与方法
1.1 材料与试剂
唑啉草酯标准品(纯度98.4%),国家农药质量监督检验中心(沈阳);唑啉草酯代谢物M2标准品(纯度99.8%),北京科润天朗环境技术有限公司;唑啉草酯代谢物M3标准品(纯度99.3%),北京科润天朗环境技术有限公司。
1.2 仪器设备
Agilent 1290/G6470型三重四极杆液质联用仪、Agilent Eclipse plus C18色谱柱(2.1 mm×50 mm×1.8μm),美国安捷伦公司;LC-30A/LCMS-8050型超高效液相色谱-三重四极杆质谱联用仪,日本岛津公司;SPS 402F型电子天平(0.01 g),梅特勒·托利多(常州)测量技术有限公司;AL204型电子天平(0.000 1 g),梅特勒·托利多国际贸易(上海)有限公司;Hei-vap型旋转蒸发仪,德国海道尔夫德祥科技有限公司;ACQUITY UPLCTMBEH C18色谱柱(2.1 mm×100 mm×1.7μm),美国沃特世科技有限公司。
供试水和土壤按相关要求采集、处理[13],主要参数信息见表1。

表1 供试水和土壤主要参数信息表
1.3 分析检测方法
1.3.1 仪器方法
色谱条件:流动相A(0.1%甲酸水溶液);流动相B(0.1%甲酸甲醇溶液);柱温为40℃;进样体积为5 μL。梯度洗脱程序见表2[12,14]。

表2 液相梯度洗脱程序
质谱条件:离子源为ESI+;检测方式为多反应监测;interface电压为4.0 kV;雾化器流速为3.0 L/min;干燥气流速为10.0 L/min;加热气流速为10.0 L/min;interface温度为300℃;DL温度为250℃;Heat Block温度为400℃。质谱信息见表3。
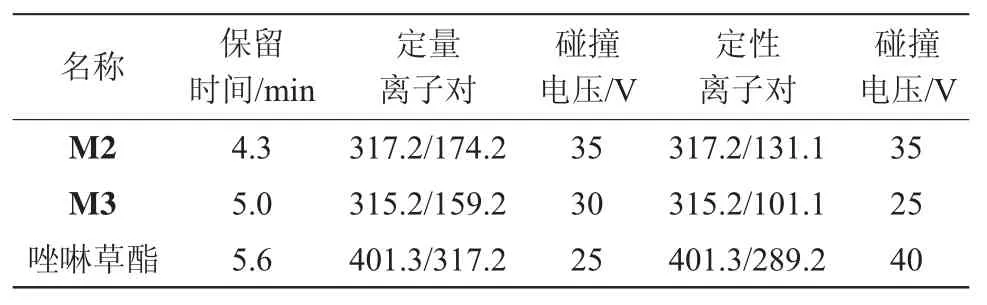
表3 农药质谱检测离子对信息
1.3.2 前处理方法
量取50 mL水样于250 mL分液漏斗中,加入10 g氯化钠,摇匀溶解,加入80 mL乙酸乙酯-二氯甲烷(1∶1,V/V,含1%0.1 mol/L盐酸),剧烈萃取3 min,上层清液过无水硫酸钠后收集于250 mL锥形瓶中,再用80、50 mL乙酸乙酯-二氯甲烷(1∶1,V/V)萃取2遍,合并上层清液并在50℃下旋蒸至近干。用5 mL乙腈-水(1∶1,V/V)定容,过膜,待LC-MS/MS检测。
称取5 g(精确至0.01 g)土壤样品于250 mL塑料离心管中,加入50 mL乙酸乙酯-乙腈(1∶1,V/V),再加入1 mL 0.1 mol/L盐酸,摇匀后超声5 min,离心,过无水硫酸钠后收集于250 mL圆底烧瓶中,再用50 mL乙酸乙酯-乙腈(1∶1,V/V)振荡提取2遍,每次30 min,随后离心,合并上清液至圆底烧瓶中,于50℃水浴中旋转蒸发至近干,冷却后用5 mL乙腈-水(1∶1,V/V)定容,过0.22μm有机相滤膜至进样瓶中,待LC-MS/MS检测。
2)几何模型建立范围为:上下游方向,以山梁山脊线为基线分别向河流上、下游方向;垂直河流方向,从河谷向山。建立有限元计算模型。
1.3.3 对照品溶液配制
分别配制唑啉草酯、唑啉草酯代谢物M2、唑啉草酯代谢物M3标准储备液(母液),然后配制1.0 mg/L的唑啉草酯、M2和M3的混合标准工作液(用色谱纯乙腈定容,2~8℃保存,有效期为1个月)。将该工作液用样品基质提取液逐级稀释,得到0.25、1.0、5.0、20、50、100μg/L的基质系列标准工作液(现配现用)。
1.4 方法验证
1.4.1 仪器方法
质谱条件:AJS-ESI+为正离子扫描;离子喷雾电压为3.5 kV;雾化器压力为310.3 kPa;干燥气温度为5 L/min;干燥气流速5 L/min;鞘气流速为11 L/min;其他质谱条件与1.3.1中相似。
1.4.2 前处理方法
与1.3.2前处理方法相同。
1.4.3 线性与范围
将1.0 mg/L的唑啉草酯、M2和M3的混合标准工作液分别用乙腈-水(1∶1,V/V)和样品基质提取液逐级稀释配制成0.25、1.0、5.0、20、50、100μg/L质量浓度的溶剂系列标准溶液和基质系列标准溶液,制作标准曲线。
1.4.4 添加回收试验
在供试自来水和红壤土中进行加标回收试验,自来水添加浓度分别为0.1、1和1 000μg/L,红壤土中添加浓度分别为1、10和1 000μg/kg,每个浓度重复6次,同时设不加药的空白对照,按上述仪器条件和样品前处理方法进行分析检测。
2 结果与分析
2.1 线性试验结果和最小检出限
当基质效应小于20%,可使用溶剂标准曲线进行计算,否则需用基质标准曲线进行计算[15]。唑啉草酯、M2、M3的基质效应在自来水中为3.17%~20.10%,红壤土中为19.8%~53.0%。验证结果存在基质效应,需选择基质标标准曲线计算。取信噪比为3倍对应的待测物质量浓度作为检出限。自来水、红壤土中唑啉草酯的线性结果见表4,相关系数均大于0.990。结果表明,该方法线性关系较好、检出限低、灵敏度高。
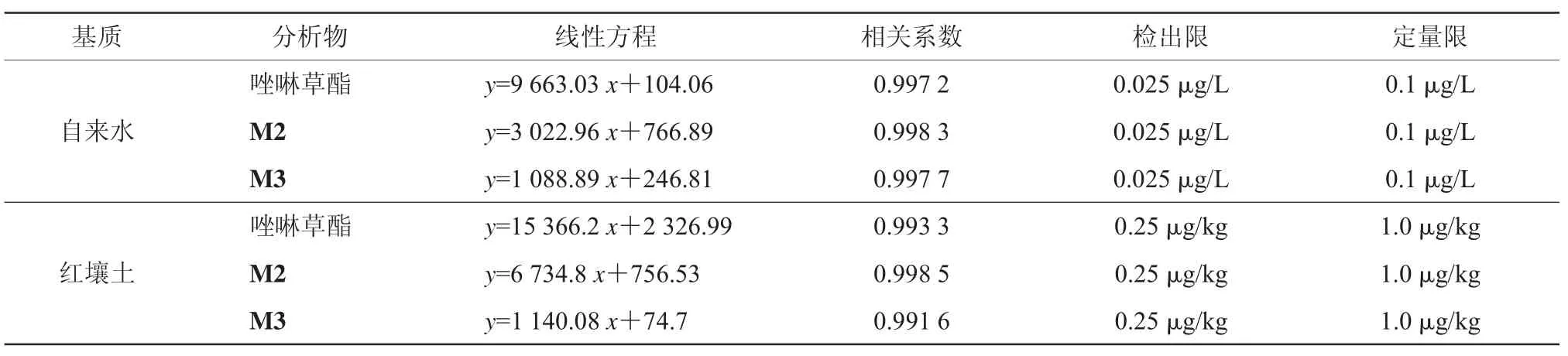
表4 不同基质中3种分析物的线性相关性、检出限和定量限(n=6)
2.2 准确度、精密度试验结果
加标回收率试验结果,见表5。在自来水和红壤土中分别添加3个水平(低、中、高)3种物质的标准混合溶液,平行测定6次,以基质标为基准计算回收率。唑啉草酯、M2、M3在自来水中添加量为0.1、1.0、1 000μg/L时,平均回收率分别为86%~98%、81%~94%、96%~105%,相对标准偏差均小于9%;在红壤土中添加量为1.0、10、1 000μg/kg时,平均回收率分别为87%~100%、91%~104%、86%~104%,相对标准偏差在均小于7%,表明该方法回收率高,准确度好。
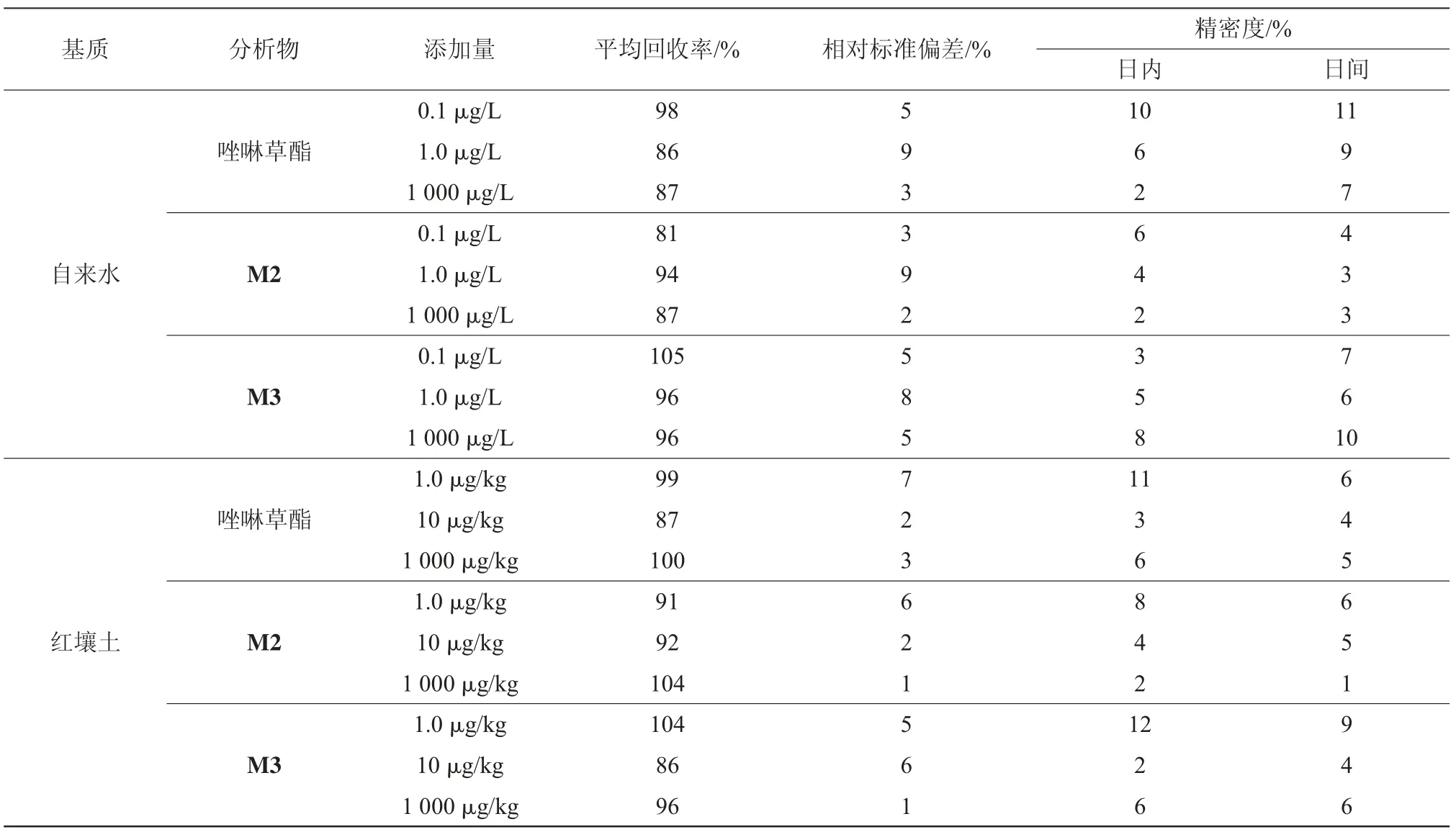
表5 自来水和红壤土中唑啉草酯、M2、M3的准确度和精密度结果(n=6)
2.3 再现性试验结果
根据相同方法进行回收率试验,自来水和红壤土中分别做了低、中、高3档添加浓度,每个浓度6次重复。结果显示,在自来水中的唑啉草酯日内和日间精密度分别为2%~10%和7%~11%;M2的日内和日间精密度分别为2%~6%和3%~4%;M3的日内和日度精密度分别为3%~8%和6%~10%。在红壤土中唑啉草酯的日内和日间精密度分别为3%~11%和4%~6%;M2的日内和日度精密度分别为2%~8%和1%~6%;M3的日内和日度精密度分别为2%~12%和6%~9%,均小于15%,表明该方法重复性好,精密度高。
2.4 选择和专属性试验结果
空白基质样品、最低档浓度标样、低档添加浓度质谱进样。结果表明,加标样品图谱中除3种标准物质峰外,未见其他杂峰产生有效干扰(图1、2),表明方法的选择性高、专属性好。

图1 自来水中唑啉草酯M2、M3子离子质谱图
2.5 耐用性试验结果
方法验证时对比了不同的仪器品牌、色谱柱型号、柱温等条件。结果显示,唑啉草酯、M2、M3含量的相对标准偏差小于1.0%,说明该方法的耐用性较好。
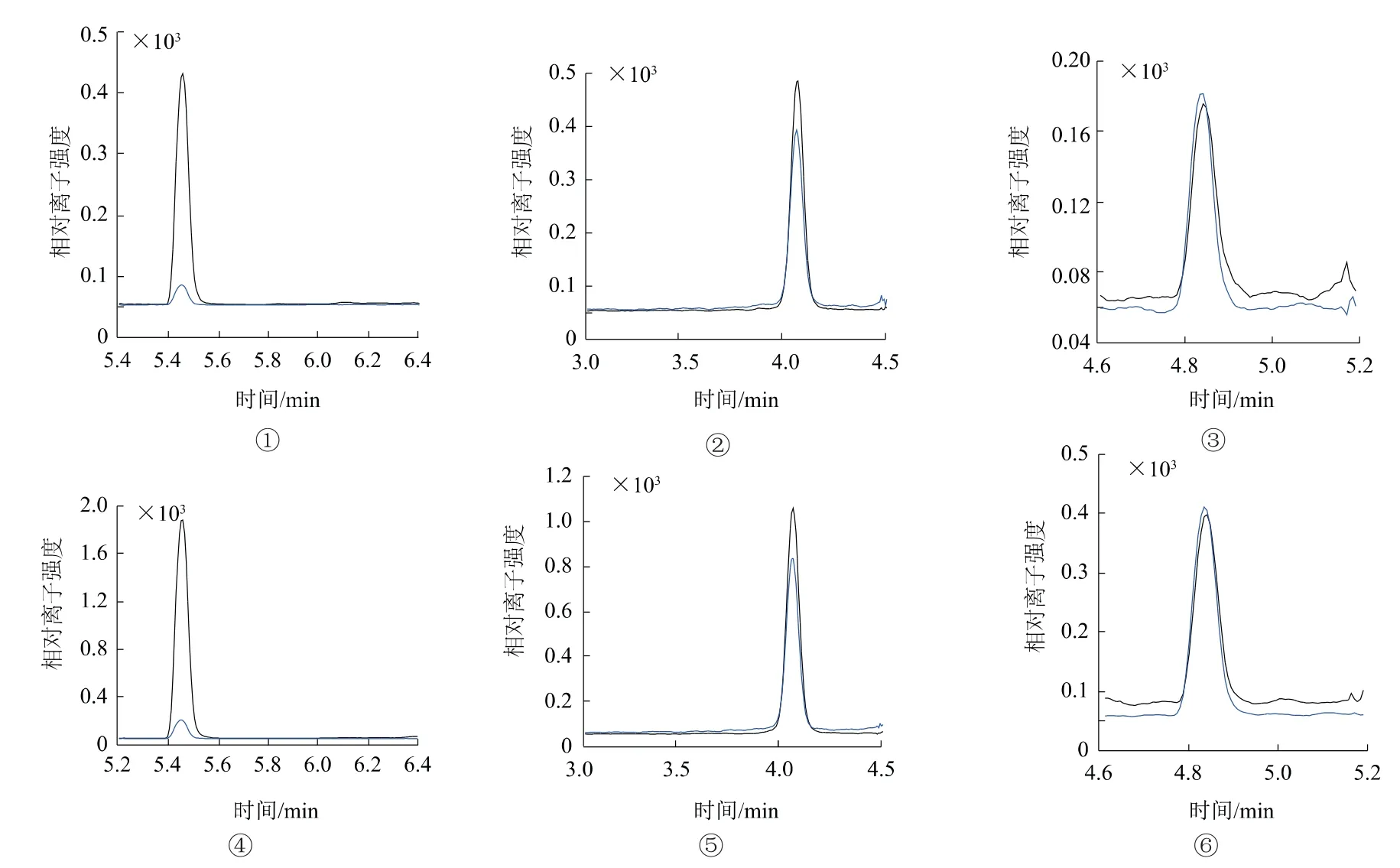
图2 红壤土中唑啉草酯M2、M3子离子质谱图
3 结 论
参照NY/T 3151—2017《农药登记土壤和水中化学农药分析方法建立和验证指南》[15],笔者采用超高效液相色谱-串联质谱法建立了自来水和红壤土中唑啉草酯、M2、M3残留量的分析方法,并对其进行了验证。该方法的灵敏度、准确度和精密度、耐用性等均符合要求,方法简单、快速、准确,一次进样能检测唑啉草酯及其代谢物的残留量,满足环境试验中痕量样品低浓度的检测要求。
猜你喜欢
现代临床医学(2022年4期)2022-09-29
中山大学学报(自然科学版)(中英文)(2022年4期)2022-08-05
系统医学(2022年6期)2022-06-13
中外葡萄与葡萄酒(2022年3期)2022-05-29
时代经贸(2019年3期)2019-11-29
科学家(2016年17期)2017-10-17
中国科技纵横(2017年15期)2017-09-09
科技视界(2016年22期)2016-10-18
分忧(2015年3期)2015-06-08
大科技·百科新说(2014年5期)2014-06-10
