
Genetic dissection of rice appearance quality and cooked rice elongation by genome-wide association study
2021-12-10 12:24:08XianjinQiuJingYangFanZhangYananNiuXiuqingZhaoCongongShenKaiChenShengTengJianlongXu
The Crop Journal 2021年6期
Xianjin Qiu,Jing Yang,Fan Zhang,Yanan Niu,Xiuqing Zhao,Congong Shen,Kai Chen,Sheng Teng*,Jianlong Xu,,*
a Engineering Research Center of Ecology and Agricultural Use of Wetland,Ministry of Education/Hubei Collaborative Innovation Center for Grain Industry,Jingzhou 434025,Hubei,China
b Laboratory of Photosynthesis and Environmental Biology,CAS Center for Excellence in Molecular Plant Sciences,Shanghai Institute of Plant Physiology and Ecology,Shanghai Institutes for Biological Sciences,Chinese Academy of Sciences,Shanghai 200032,China
c National Key Facility for Crop Gene Resources and Genetic Improvement,Institute of Crop Sciences,Chinese Academy of Agricultural Sciences,Beijing 100081,China
d Shenzhen Branch,Guangdong Laboratory for Lingnan Modern Agriculture,Genome Analysis Laboratory of the Ministry of Agriculture,Agricultural Genomics Institute at Shenzhen,Chinese Academy of Agricultural Sciences,Shenzhen 518120,Guangdong,China
e University of Chinese Academy of Sciences,Beijing 100049,China
Keywords:
A B S T R A C T Appearance and cooked rice elongation are key quality traits of rice.Although some QTL for these traits have been identified,understanding of the genetic relationship between them remains limited.In the present study,large phenotypic variation was observed in 760 accessions from the 3K Rice Genomes Project for both appearance quality and cooked rice elongation.Most component traits of appearance quality and cooked rice elongation showed significant pairwise correlations,but a low correlation was found between appearance quality and cooked rice elongation.A genome-wide association study identified 74 QTL distributed on all 12 chromosomes for grain length,grain width,length to width ratio,degree of endosperm with chalkiness,rice elongation difference,and elongation index.Thirteen regions containing QTL stably expressed in multiple environments and/or exerting pleiotropic effects on multiple traits were detected.By gene-based association analysis and haplotype analysis,46 candidate genes,including five cloned genes,and 49 favorable alleles were identified for these 13 QTL.The effect of the candidate gene Wx on rice elongation difference was validated by a transgenic strategy.These results shed light on the genetic bases of appearance quality and cooked rice elongation and provide gene resources for improving rice quality by molecular breeding.
1.Introduction
Rice(Oryza sativaL.)is the staple food of more than half of the world’s population.With improvement of living standards in riceconsuming regions,grain quality is receiving attention from both consumers and producers and has become a key rice breeding target.Grain quality may be divided into milling,appearance,cooking and eating,and nutritional quality[1].Appearance quality is composed of grain shape and grain chalkiness.Grain shape consists of grain length(GL),grain width(GW),and length-to-width ratio(LWR).The price of rice with different grain shapes(as expressed in dehulled seeds)varies by region.Rice with a long and slender shape is preferred in southern China,southern and southeastern Asia,and the USA,whereas short and round-grain rice is preferred in northern China,Democratic People’s Republic of Korea,Republic of Korea,and Japan.Because grain shape also influences milling quality and grain weight[2],it affects yield.Grain chalkiness includes white back,white core,and white belly based on the parts that become chalky,and is an undesirable trait.It is usually evaluated by percentage of grains with chalkiness and degree of endosperm with chalkiness(DEC).Cooked rice elongation,evaluated by rice elongation difference(RED)and rice elongation index(REI),strongly influences eating and cooking quality,and elongation in the longitudinal direction and nearly no elongation in the lateral direction are considered to indicate good quality and are well received in the international rice market[3].
All appearance-quality traits are typical quantitative traits,controlled by multiple genes and affected by environment[4].With the development of rice functional genomics and DNA marker technology,numerous QTL have been identified on all 12 chromosomes in various mapping populations(http://www.gramene.org/).Many genes for grain shape have been cloned,includingGW2,GW5,GS3,GS5,GL3.1,GS2,GL7/GW7/SLG7,GLW7,SGDP7,GS9,smg11,OsMADS1,andOsSNB[5].Some genes for chalkiness have been cloned,includingOsPPDKB,SSIIIa,GIF1,ms-h,FLO2,OsRab5a,Chalk5[1],andPFP1[6].Some QTL,includingqGRL1.1[7]for grain shape andqPGWC-7[8]andqPGWC-8[9]for grain chalkiness,have been fine-mapped.
Cooked rice elongation,a quantitative trait,receives close attention in the international rice market.Because the inheritance pattern of cooked rice elongation is inconsistent in different crosses,it is difficult to elucidate its genetic basis,which has remained ambiguous.Basmati rice is a desirable type that shows high elongation without width increase upon cooking[10].Recently,three QTL were identified by a QTL-seq strategy using an F2population derived from a cross between Basmati and a Thai fragrant rice variety[11].Two were located near starch-synthesis genes.Some QTL for rice elongation have been identified in non-Basmati cultivars[3,12,13].
In recent years,a new approach named genome-wide association study(GWAS)based on large germplasm collections and linkage disequilibrium(LD)has been used to identify rice QTL governing complex traits.Using this strategy,genomic regions associated with rice appearance quality have been identified.Seven QTL for grain length and width were identified in 517 landraces by re-sequencing[14].Zhao et al.[15]identified 56 QTL for grain shape of rice seed and brown rice respectively in 413 diverse inbred accessions using 44K SNP genotypes.Twenty-six QTL associated with appearance quality were identified in 272xian(indica)accessions genotyped for 22,266 SNPs[16].Fifty-three QTL and 19 candidate genes for rice appearance quality were identified using 258 accessions from the 3K Rice Genomes Project(RGP)using high-through genotyping[17].Qiu et al.[18]identified 70 QTL for grain shape using 1016 accessions.
In the present study,the appearance quality and cooked rice elongation of a diverse panel containing 760 accessions selected from the 3K RGP[19]were evaluated.GWAS was performed to identify QTL for rice appearance quality and cooked rice elongation using high-quality SNPs generated from 3K RGP by highthroughput sequencing.In some important QTL regions,genebased association analysis was performed using all available SNPs from the RFGB 2.0 database[20].Haplotype analysis was performed to identify candidate genes and favorable alleles.The association results shed light on the genetic basis of rice appearance quality and cooked rice elongation and provide genes and favorable alleles for improving rice quality by molecular breeding.
2.Material and methods
2.1.An association population
A total of 760 accessions from 3K RGP were used.They were from 62 countries and regions(Table S1),and consisted of five types,includingxian(indica,416),geng(japonica,284),aus(31),admixture(19),andaroma(10).
2.2.Field experiment and trait evaluation
All accessions were planted in the winter seasons of 2015,2016,and 2017 at the Station of the Institute of Crop Sciences,Chinese Academy of Agriculture Science at Sanya,Hainan province.Field experiments were conducted using a randomized complete block design with two replications.Each plot contained three rows of 10 plants each at a spacing of 20 cm between plants and 25 cm between rows.All field management followed local farmers’practices.
At the mature stage,eight plants in the second row were bulkharvested.Seeds were dried at room temperature for three months and used to evaluate GL,GW,and LWR in 2015,2016,and 2017,DEC in 2015 and 2016,and RED and REI in 2016.GL,GW,LWR,and DEC were measured following Qiu et al.[21].For REI and RED,10 unbroken milled rice were randomly selected and their lengths were recorded.They were soaked in 7.5 mL distilled water for 0.5 h,boiled for 10 min,and cooled and dried for 3 h at room temperature and their lengths were recorded again.The mean length was used for further analysis.RED was calculated as cooked rice length minus milled rice length and REI was calculated as the percentage of RED divided by milled rice length.Phenotypic data are presented in Table S2.
2.3.Population structure and linkage disequilibrium decay
The 3K RGP 4.8 M SNP dataset was retrieved from the Rice SNPSeek Database(http://snp-seek.irri.org/)[22].To avoid the influence of linked SNPs during the population structure analysis,the LD pruning tool of PLINK 1.9[23]was used to obtain a subset of 73,162 independent SNPs with MAF>5% and a missing data ratio<0.1 using the parameter values‘‘indep-pairwise 50 10 0.1”.The genetic structure of the whole population was predicted with the ADMIXTURE program[24].GCTA software was used to conduct a principal component analysis to estimate the number of subpopulations in the GWAS panel.Genomic LD decay was estimated based on the coefficients of determination(r2)between all pairs of loci using PopLDdecay 3.30[25]in a 500 kb distance.
2.4.GWAS
GWAS was performed to detect trait–SNP associations for appearance quality and cooked rice elongation using 488,973 high-quality SNPs and the trait values of the 760 accessions.The SVS software package(SNP & Variation Suite,version 8.4.0)was used for GWAS analysis[18].The EMMAX(Efficient Mixed-Model Association eXpedited)implementation of the single-locus mixed linear model was fitted to the marker dataset.Significant SNPs affecting the investigated traits were claimed when the test statistics reachedP<1.0×10-5.Because the LD decay of the 760 accessions was 141 kb,peak SNPs within 141 kb were combined into a single QTL.
2.5.Candidate gene identification and haplotype analysis
Gene-based association analysis and haplotype analysis were performed to identify candidate genes and favorable alleles for important QTL.Favorable alleles for each trait were designated as alleles with increasing effects for GL,GW,RLW,RED,and REI or decreasing effect for DEC.QTL identified in two or three years and/or affecting more than two traits and containing cloned genes or not previously reported were assigned as important QTL.For gene-based association mapping,QTL with peak SNP having the lowestPvalues were used for analysis.Three steps were conducted to identify QTL candidate genes.First,all genes located within 70 kb upstream and downstream of the peak SNP of each important QTL were retrieved from the Rice Genome Annotation Project(http://rice.plantbiology.msu.edu/).Next,all available SNPs located within these genes(ignoring SNPs located in intergenic regions)were retrieved from 32 M SNPs data generated from 3K RGP in the RFGB 2.0 database[20]and used to perform association analyses.The threshold was defined as 1.0×10-5(-log10(P)=5).Finally,all SNPs in coding regions and 5′flanking sequences of genes(<2 kb from the first TAG)with-log10(P)above the threshold were used for identifying haplotypes.Candidate genes were assigned by testing for significant differences for all detected traits among major haplotypes(containing more than 10 samples)by ANOVA with a threshold ofP<0.001.For QTL regions containing cloned genes,the cloned genes were considered the only candidate genes for haplotype analysis.For cloned or most likely candidate genes of important QTL regions,two-tailed Fisher’s exact test was performed to determine significant differences in frequencies of gene haplotypes betweenxianandgenggroups using the‘fisher.test’function in R(http://www.r-project.org/),using a threshold ofP<0.01.
2.6.Validation of Wx for RED
LOC_Os06g04169andWxwere identified as candidate genes for RED in the region 1.63–1.83 Mb on chromosome 6.To validate the effect ofWxon RED,the full-length genomic DNA sequence ofWx(including the promoter and the codon region)of CX114 was amplified and cloned into the binary vector pCAMBIA1301 and then transformed into Nipponbare.At the T1generation,10 homozygous positive transgenic plants were evaluated for RED as described above,and 10 negative transgenic plants were used as control.At-test was used to compare their mean RED values.
3.Results
3.1.Population structure and LD pattern
Based on the previous neighbor-joining tree of 3K RGP,the 760 accessions were derived from all 12 rice subpopulations(Fig.S1A).To reassess population structure among all 760 accessions sampled,admixture analysis and principal component analysis(PCA)were performed in the GWAS panel.Similar results for the population structure analysis by ADMIXTURE and PCA were observed in the 760 accessions compared with the whole 3K RGP(Fig.S1B and C).LD decayed rapidly with the increase of physical distance between SNP pairs and reached its half maximum within 141 kb for the whole GWAS panel(Fig.S1D),a pattern of LD decay similar to that of the 3K RGP.
3.2.Trait values and correlations in the association population
Appearance quality and cooked elongation phenotypes are described in Table 1.A wide range of variation was observed for all evaluated traits(with CVs ranging from 11.74% to 91.87%),and DEC,RED,and REI varied more widely than grain shape traits(with CVs>20% for DEC,RED,and REI and<20% for GL,GW,and LWR)(Table 1).All traits appeared to be normally distributed in all years,suggesting that they were controlled by multiple genes.GL,GW,and DEC differed among years.GL and GW of all accessions were greater in 2017 than in 2015 and 2016,and their DEC was lower in 2015 than in 2016(Table 1).
Pairwise correlations among traits were similar across years(Table 2).Low to high correlations were detected for appearancequality traits,whereas low correlations were found between appearance quality and cooked rice elongation.GL was lowly and negatively correlated with GW and DEC,and moderately and positively correlated with LWR.GW showed moderate negative correlation with LWR and moderate positive correlation with DEC.LWR was highly and negatively correlated with DEC.These results suggested that rice with longer grain would have smaller GW,higher LWR,and lower DEC.RED was highly and positively correlated with REI,lowly and positively correlated with GW and DEC,and lowly and negatively correlated with LWR.REI showed low and positive correlations with GW and DEC,and moderate and negative correlations with GL and LWR,indicating that rice with wide and short grains and high DEC would elongate more after cooking.
3.3.Detection of QTL by GWAS
In total,74 QTL were identified for five appearance-quality and two cooked rice elongation traits,ranging from six for RED to 23 for GW.Among them,18 QTL were stably expressed in all three years,9 QTL were detected in two of the three years,and 47 QTL were identified in only one year of 2015,2016,and 2017(Table 3;Fig.S2).
For GL,14 QTL were identified.Among them,5 QTL(qGL3.1,qGL5.1,qGL6.1,qGL7.1,andqGL7.2)were stably expressed in all three years and explained mean phenotypic variation of 46.3%,17.9%,0.8%,6.4%,and 6.9%,respectively.qGL8was identified in both 2015 and 2016 and accounted for 7.1% and 15.2% of phenotypic variation.The remaining eight QTL were identified in only one year and accounted for 1.3%–8.5% of phenotypic variation.
Twenty-three QTL were detected for GW.Among them,nine(qGW3.5,qGW4.2,qGW5.1,qGW5.2,qGW7.1,qGW7.3,qGW9,
qGW11andqGW12.1)were stably expressed in all three years and accounted for mean variation of 1.3%–32.7%.Five QTL,qGW2,qGW3.3,qGW4.3,qGW6.1,andqGW7.2,were detected in two years of 2015,2016 and 2017 with mean percentage of phenotypic variation of 1.2%–25.5%.The remaining nine QTL were detected in only one year and accounted for 1.1%–28.2% of phenotypic variation.
For LWR,11 QTL were found on chromosomes 1,2,3,5,6,7,and 10.Four QTL,qLWR3.2,qLWR5.2,qLWR7.1,andqLWR7.2,were found in all three years and explained 6.2%–39.8% of phenotypic variation.Three QTL,qLWR1,qLWR3.1andqLWR5.1commonly detected in two years with mean percentage of phenotypic variation of 10.8%,19.5%,and 5.6%,respectively.The remaining four QTL were identified only in either 2016 or 2017,and explained 1.2%–9.6% of phenotypic variation.
Twelve QTL for DEC were identified in 2015 and 2016.None was found in both years.Five QTL(qDEC1,qDEC3,qDEC5.2,qDEC8.2,andqDEC9)were identified only in 2015 with the percentage of phenotypic variation of 1.6%–22.7%.Seven QTL includingqDEC4,qDEC5.1,qDEC7,qDEC8.1,qDEC8.3,qDEC10,andqDEC11were detected only in 2016 with explained percentages of phenotypic variation of 1.1%–14.8%.
For RED in 2016,six QTL were detected on chromosomes 6,9,10 and 11.They explained 3.1%–5.7% of phenotypic variation.
For REI in 2016,eight QTL were identified on chromosomes 1,5,6,9,and 11 in 2016,accounting for 1.1%–6.0% of phenotypic variations.
3.4.Candidate gene analyses of important QTL
Among the 74 QTL,13 QTL regions consistently detected in different years or pleiotropic for different traits and containing cloned genes or newly identified QTL were assigned as important QTL.Gene-based association analyses were performed using all SNPs in genes within 100 kb to either side of the peak SNP for these QTL regions.A total of 46 candidate genes including five cloned genes were identified(Figs.1,2,S3;Table 4).
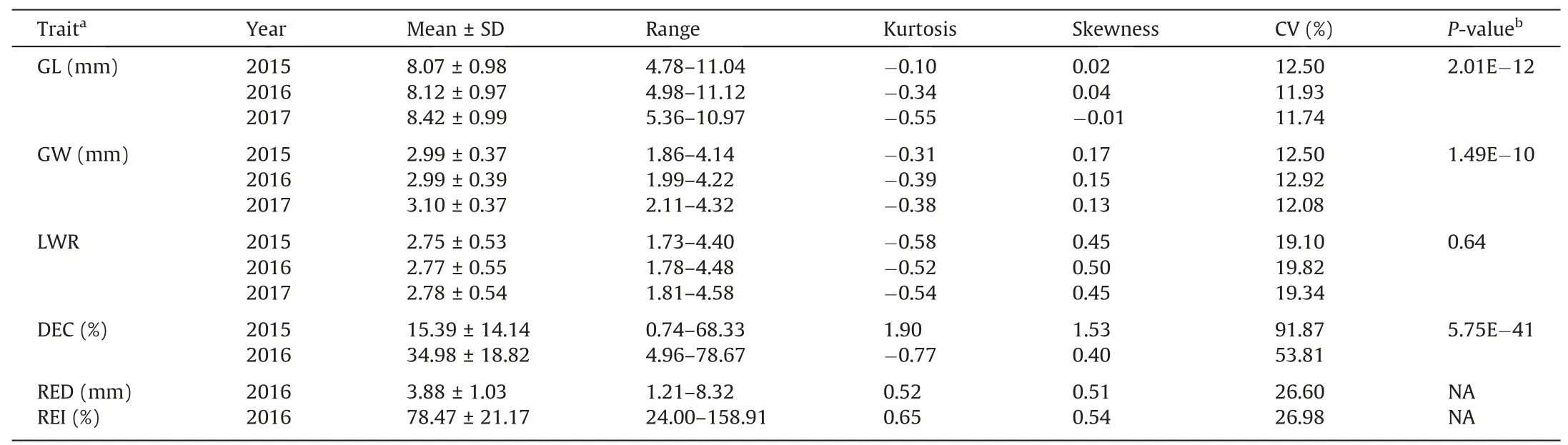
Table 1Appearance quality and cooked rice elongation in 760 rice accessions.

Table 2Correlation coefficients of appearance quality and cooked elongation measured in the 760 rice accessions.
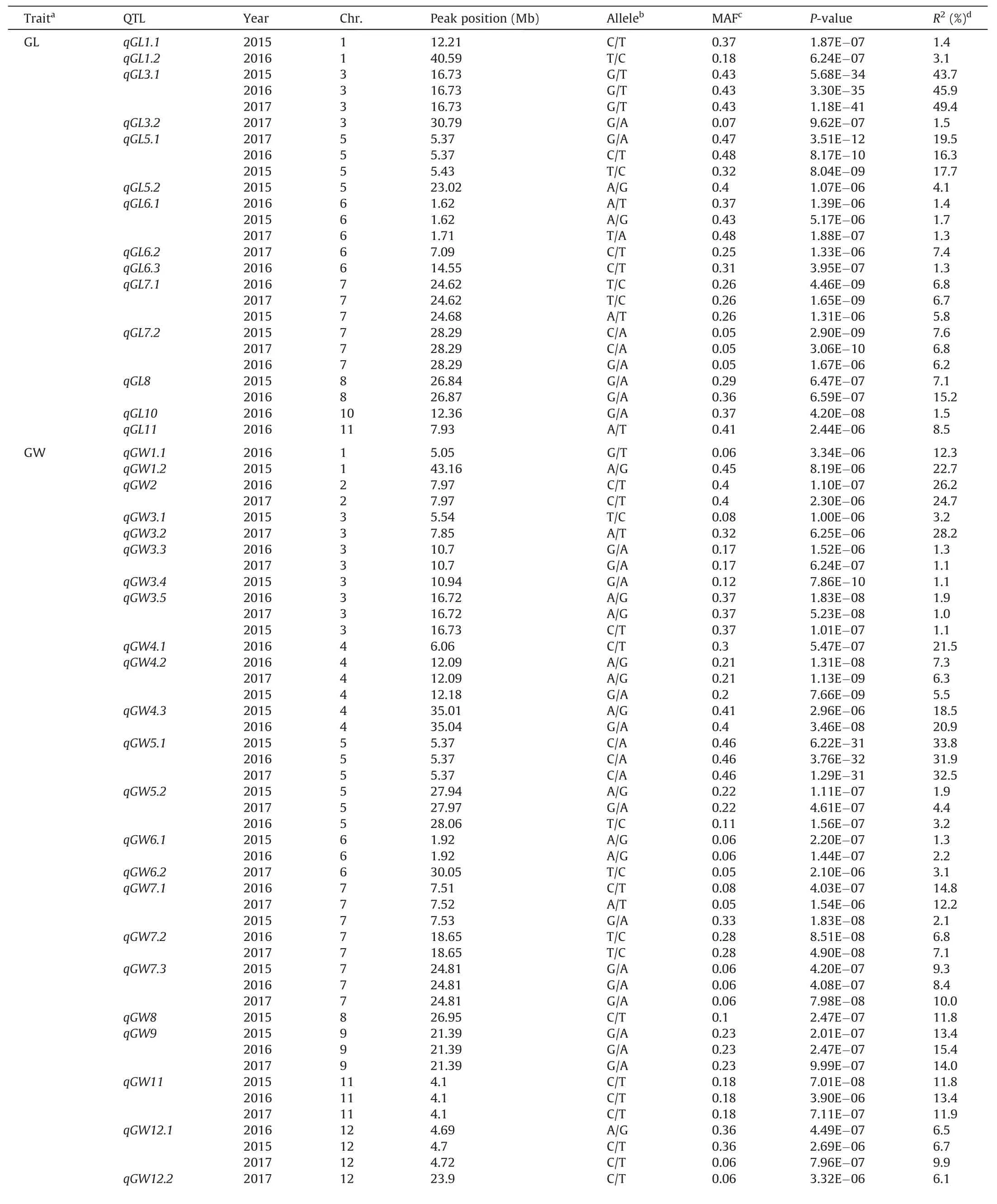
Table 3QTL for appearance quality and cooked rice elongation in 760 rice accessions by GWAS.

Table 3(continued)
There were five QTL regions with cloned genes.In the region of 16.63–16.83 Mb on chromosome 3 harboringGS3,five haplotypes were identified based on 46 SNPs with–log10(P)>5.Hap 4 and Hap 5 were associated with higher GL,lower GW,and higher LWR.
ForqLWR5.2in the region of 5.27–5.47 Mb on chromosome 5 containingqSW5/GW5/GSE5,using five significant SNPs,six haplotypes were identified,and Hap 5 and Hap 6 had higher GL,lowerGW,higher GLW,lower DEC,and lower REI than Hap 1,Hap 2,and Hap 4.
qGW7.1was identified in the region of 7.43–7.63 Mb of chromosome 7 containing a cloned gene(OsSNB).GW was significantly different between two haplotypes identified based on nine significant SNPs.
In the region of 24.64–24.84 Mb on chromosome 7 harboringGL7/GW7/SLG7,two haplotypes were identified based on three significant SNPs,and significant phenotypic differences of GL and GW were detected between them.
qGW8was identified in the region of 26.80–27.00 Mb of chromosome 8 and contained a cloned gene,WTG1.Four haplotypes were identified based on 10 significant SNPs.Hap 4 was associated with longer GL and wider GW.
In the remaining 8 QTL regions,no cloned gene was present.ForqGW4.2in the region 11.98–12.18 Mb on chromosome 4,388 SNPs in 16 genes were used for gene-based association analysis.Only one gene(LOC_Os04g21390)contained one SNP with–log10(P)>5,and two haplotypes were identified.Hap 2 had higher GW than Hap 1.
In the region 0–0.15 Mb on chromosome 5 forqLWR5.1,308 SNPs of 25 genes were used for gene-based association analysis.SNPs with–log10(P)>5 were distributed around two candidate genes,LOC_Os05g01020andLOC_Os05g01040.There were two haplotypes for each of them,and there was a significant difference for LWR between the two haplotypes for both genes.
qGW5.2was detected in the region 27.84–28.04 Mb on chromosome 5,in which region 422 SNPs in 28 genes were used for association analysis.Three SNPs,one in each of three genes(LOC_Os05g48740,LOC_Os05g48800,andLOC_Os05g48820),had significantPvalues(–log10(P)>5).All three candidate genes had two haplotypes,and GW was significantly different between them for all candidate genes.
A total of 209 SNPs of 25 genes were used for gene-based association analysis in the region 1.63–1.83 Mb on chromosome 6.LOC_Os06g04169andLOC_Os06g04200(Wx)containing SNPs with–log10(P)>5 were assigned as candidate genes.There were significant differences for GL,GW,and RED between two haplotypes ofLOC_Os06g04169.ForLOC_Os06g04200(Wx),three haplotypes were identified based on two significant SNPs.Hap 1 and Hap 2 had lower GW and higher RED than Hap 3.
TheqREI6.4was detected in the region 26.17–26.37 Mb on chromosome 6 harboring 654 SNPs of 26 genes.SNPs with–log10(P)>5 hits three candidate genes:LOC_Os06g43670,LOC_Os06g43680,andLOC_Os06g43710.Two,one,and one SNPs divided these genes into two haplotypes,and there were significant differences for RED and REI between the two haplotypes for all three genes.
qGW7.2was identified in the region of 18.55–18.75 Mb of chromosome 7 harboring 504 SNPs of 22 genes.Of these,eight genes harbored significant SNPs with two haplotypes.Haplotype analysis suggested that there were significant differences between two haplotypes for both GW and LWR for all eight candidate genes.
ForqGL7.2in the region 28.19–28.39 Mb on chromosome 7,291 SNPs of 33 genes were used for gene-based association analysis.LOC_Os07g47160,LOC_Os07g47170,LOC_Os07g47230,LOC_Os07g47240,LOC_Os07g47320,LOC_Os07g47340, andLOC_Os07g47350were assigned as candidate genes.Haplotype analysis showed significant differences between two haplotypes for GL in all of them.
qGW12.1was predicted in the region 4.59–4.79 Mb on chromosome 12 covering 371 SNPs of 22 genes.Fifteen genes harboring significant SNPs were found.All haplotypes showed significant differences within all candidate genes for GW.
3.5.Wx affected RED
In the region 1.63–1.83 Mb on chromosome 6,two candidate genes were identified for RED:LOC_Os06g04169andLOC_Os06g04200(Wx)(Figs.1,2A,S3I;Table 4).In previous studies,many QTL for rice elongation were detected in this region[3,12,13].Moreover,starch synthetic genes were thought to be important for rice elongation[11].We accordingly consideredWxthe most likely candidate gene for RED.To determine whetherWxaffected RED,the whole gene of CX114(Guangluai 4,Hap 2)was introduced into Nipponbare(Hap 3)background by transgenic strategy.The RED of Guangluai was 4.48 mm,significantly higher than that of Nipponbare.At the T1generation,10 plants homozygous for the positive allele and 10 homozygous for the negative allele were evaluated for RED.The RED of the positive plants was 4.35 mm,significantly higher than that of the negative plants(3.20 mm)(Fig.2B).This result confirmed thatWxaffected RED and that Hap 2 had higher RED than Hap 3.
4.Discussion
4.1.Genetic overlap between appearance quality and cooked rice elongation
Previous studies reported large number of genetic overlap among rice traits such as yield[26,27],appearance quality[21,28],heavy metal tolerance[29],and salt tolerance[30].In the present study,although some appearance-quality traits showed significant correlations with rice elongation traits(Table 2),all the correlation coefficients were below 0.40,suggesting that appearance quality and cooked rice elongation had only slight genetic overlap.Moreover,among 60 appearance-quality QTL and 14 cooked rice elongation QTL,only two regions,5.27–5.47 Mb on chromosome 5 and 1.63–1.83 Mb on chromosome 6(10.0%for appearance quality QTL and 21.4% for cooked rice elongation QTL)showed pleiotropic effects on both appearance quality and cooked rice elongation(Table 3;Fig.S3).This finding was in agreement with the previous reports of no genetic overlap between them[12].Within appearance quality traits,grain shape and DEC also showed low genetic overlap.Only the region 5.27–5.47 Mb on chromosome 5 showed a pleiotropic effect on both grain shape traits and DEC.This region contained three cloned genes for GW and chalkiness that were tightly linked with one another,qSW5/GW5/GSE5[31],GS5[32],andChalk5[33].It was consistent with our previous studies[16].Thus,appearance quality and cooked rice elongation,and grain shape and DEC could be simultaneously improved by molecular breeding without fear of genetic drag.
4.2.Comparison of important QTL regions in the present study with previously reported QTL
Thirteen important QTL regions were identified for four traits of appearance quality and two traits of cooked rice elongation(Figs.1,S3;Table 4).In contrast to previous studies,most regions were detected in the same or adjacent regions of cloned genes.For instance,qGL3.1,qGW3.5,andqLWR3.2for grain shape were mapped together withGS3[34];qGL5.1,qGW5.1,qLWR5.2,qDEC5.2,andqREI5were mapped together withqSW5/GW5/GSE5[31]and nearChalk5[33];qRED6.1andqREI6.1were identified together withWx[35];qGW7.1was consistent withOsSNB[36];qGL7.1,qGW7.3,andqLWR7.2were mapped together withGL7/GW7/SLG7[37];qGL8andqGW8were mapped together withWTG1[38].qLWR5.1was in the region adjacent toGS5[32];qGW7.2andqLWR7.1were located in the region adjacent toGLW7[39],andqGL7.2was detected nearSGDP7[40].

Fig.1.Manhattan plots of gene-based association analysis in 13 important QTL regions.
The remaining four important regions were identified with no cloned genes near them.However,there were many QTL mapping together or in adjacent regions.For instance,qGW4.1andqGW5.2were identified in regions adjacent toqGW4.1[16]andqGW5.3[18],respectively;qGW12.1was detected nearkw12.1[41].Allelic correspondences of these QTL for appearance quality and cooked rice elongation identified in the present study with previously reported QTL should be further investigated by fine mapping and cloning.
4.3.Candidate gene identification for important QTL

Fig.2.Haplotype analysis and transgenic validation of Wx.
Combining GWAS and gene-based association mapping with haplotype analysis yielded 46 candidate genes responsible for 13 important QTL regions(Figs.1,S3;Table 4).Among them,five cloned genes were associated with appearance quality.Besides these,there were 41 candidate genes for eight important QTL.ForqGW4.2there was only one candidate gene,LOC_Os04g21390,which encodes a retrotransposon protein.Of the two candidate genes ofqLWR5.1,LOC_Os05g01040encoding a serine/threonineprotein kinase is the most likely candidate gene,given that a cloned gene,GS5,encodes a serine carboxypeptidase and is a positive regulator of grain size[32].The expression level ofGS5was significantly associated with grain width.Another cloned gene,MIS2,encoding receptor-like kinase,also controlled GL and GW[42].It is highly expressed in developing panicles,especially at the 15-cm length stage.Moreover,LOC_Os05g01040is highly expressed in panicles in the early stage of panicle development(https://ricexpro.dna.affrc.go.jp/).In the 27.84–28.04 Mb interval on chromosome 5,three candidate genes were suggested.Among them,LOC_Os05g48800encoding drought-induced 19 protein was the most likely.Drought-induced protein Di19-3 interacted with an IAA protein,AtlIAA14,and participated in the IAA pathway[43].Given that a previous study[5]suggested that IAA plays an important role in regulating grain size,it is possible thatLOC_Os05g48800controls GL and GW in rice via the IAA pathway.Of three candidate genes forqREI6.4,LOC_Os06g43710encodes glucosidase II beta subunit-like domain containing protein.Glucosidase is an enzyme that hydrolyzes the glucuronide glycoside bond and releases glucose,functioning in sugar metabolism[44].Starch synthetic genes were reported to be important for rice elongation[11].Accordingly,LOC_Os06g43710was assigned as the most likely candidate gene forqREI6.4.Of eight candidate genes governingqGW7.2,LOC_Os07g31450(CRH729/NAAL1)was the most likely.It was expressed in all organs at developing stage[45].A mutant ofCRH729/NAAL1showed decreased plant height,panicle length,flag leaf length and width,root number,and length and GL.In the region ofqGL7.2governing seven candidate genes for GL,LOC_Os07g47160(OsFBX259)encodes an F-box domaincontaining protein.Its homologous gene,OsFBX72,also encodes a F-box domain-containing protein and could control GL and GW[46].It was expressed in all organs,and its encoded protein interacted with Skp1.Thus,LOC_Os07g47160is the most likely candidate gene forqGL7.2.Among 15 candidates ofqGW12.1,the most likely one wasLOC_Os12g09089.Similar toLOC_Os05g01040forqLWR5.1,it also encodes a protein kinase.Accordingly,LOC_Os04g21390,LOC_Os05g01040,LOC_Os05g48800,LOC_Os06g43710,LOC_Os07g31450,LOC_Os07g47160, andLOC_Os12g09089were assigned as the most likely candidate genes for newly identified genomic regions associated with grain appearance quality and rice elongation.Transgenic validation is needed to verify their functions and genetic pathways for grain appearance quality and rice elongation.
Significant differences in allele frequency between thexianandgenggroups were observed for all of the above candidate genes butLOC_Os07g47160(Table S3).For favorable alleles,the frequency of Hap 1 ofLOC_Os07g31450,Hap 2 ofLOC_Os05g01040andLOC_Os06g04200(Wx),Hap 4 ofGS3,and Hap 5 ofqSW5/GW5/GSE5were significantly higher in thexiangroup than in thegenggroup,indicating that they have been used mainly inxianquality breeding.In contrast,the frequency of Hap 1 ofGL7/GW7/SLG7andLOC_Os12g09089,Hap 2 ofOsSNB,LOC_Os04g21390,LOC_Os05g48800,andLOC_Os06g43710,Hap 4 ofWTG1,and Hap 6 ofqSW5/GW5/GSE5were significantly higher in thegenggroup than in thexiangroup,indicating that they have been used mainly in quality breeding ofgengrice.
4.4.Application in molecular breeding for improving rice appearance quality and cooked rice elongation
Improving rice quality,especially appearance quality and cooked rice elongation,is a key target in rice breeding[21,47].In the present study,appearance-quality traits were significantly correlated with cooked rice elongation traits(Table 2).This phenomenon could be explained by the presence of pleiotropic QTL(Fig.S3).Among 13 important QTL regions,six were identified for two to four appearance quality traits,and one region(26.17–26.37 Mb on chromosome 6)showed effects on both RED and REI.However,the genetic overlap between them was low.Thus,appearance quality and rice elongation could be simultaneously improved.Of 13 important QTL regions,12 showed large effects on grain shape.A total of 84 elite accessions with long grain(GL>9.0)and high LWR(LWR>3.0)were screened(Table S4).All of them carried combinations of 3–11 favorable alleles,and a breeder could pyramid different haplotypes using the above elite accessions as donor parents for improving grain shape.qSW5/GW5/GSE5ofqLWR5.2strongly affected DEC,and 15 elite accessions with DEC<10%were selected(Table S5).Half of them carried favorable alleles at the locus.We can simultaneously improve grain shape and chalkiness by pyramiding favorable alleles ofqSW5/GW5/GSE5and the above novel alleles for grain shape.Among the above elite accessions,IRIS_313-8173,carrying almost all favorable alleles,had a large value as a donor parent for improving grain appearance quality.Finally,three important QTL regions were identified for rice elongation.A total of 97 elite accessions with high RED and REI(RED>4.4 mm and REI>100%)were identified and could be used as donors for improving rice elongation(Table S6).Most of them carried one to three favorable alleles.A breeder could pyramid suitable favorable alleles of the 13 important QTL regions using the above elite accessions as donors to breed rice cultivars with excellent appearance quality and elongation using marker-assisted selection.Dozens of SNPs for the above candidate genes were identified and could be easily converted to Kompetitive Allele Specific PCR(KASP)markers for pyramiding multiple QTL or genes by marker-assisted selection.
5.Conclusions
A total of 74 QTL for rice appearance quality and cooked rice elongation were identified by GWAS in 760 rice accessions.Thirteen important QTL regions were detected for more than two traits or stably expressed in multiple environments.Forty-six candidate genes and 49 favorable alleles were identified based on gene-based association analysis and haplotype analysis.LOC_Os06g04200(Wx)was validated as a functional gene underlying RED with Hap 2 as a favorable allele.These results will enrich the genetic bases of appearance quality and cooked rice elongation and provide gene resources for improving rice quality by molecular breeding.
CRediT authorship contribution statement
Jianlong Xu:Project administration and Writing-review &editing.Sheng Teng:Project administration and Writing-review& editing.Jing Yang:Investigation.Yanan Niu:Investigation.
Xiuqing Zhao:Investigation.Congcong Shen:Investigation.Kai Chen:Investigation.Xianjin Qiu:Writing-original draft,Data curation and Formal analysis.Fan Zhang:Data curation and Formal analysis.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was funded by the National Key Research and Development Program of China(2016YFD0100301),Project for Cultivating New Transgenic Varieties(2016ZX08009003-004),the Agricultural Science and Technology Innovation Program and the Cooperation and Innovation Mission(CAAS-ZDXT202001),Open Fund of Hubei Collaborative Innovation Center for Grain Industry(HCICGI2020-06),and the National Natural Science Foundation of China(U19A2025 and 31870229).
Appendix A.Supplementary data
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2020.12.010.

- The Crop Journal的其它文章
- WheatGene:A genomics database for common wheat and its related species
- A gain-of-function mutation of OsMAPK6 leads to long grain in rice
- The importance of aboveground and belowground interspecific interactions in determining crop growth and advantages ofpeanut/maize intercropping
- Mining favorable alleles for five agronomic traits from the elite rapeseed cultivar Zhongshuang 11 by QTL mapping and integration
- Genetic gains with genomic versus phenotypic selection for drought and waterlogging tolerance in tropical maize(Zea mays L.)
- BSA-seq-based identification of a major additive plant height QTL with an effect equivalent to that of Semi-dwarf 1 in a large rice F2 population