
Immunostimulatory role of rBmHSP60 from filarial parasite Brugia malayi
2021-12-10 06:21VikasKushwahaSukhbirKaur
Vikas Kushwaha, Sukhbir Kaur
Leishmania Research Laboratory, Department of Zoology, Panjab University, Sector-14, Chandigarh 160014, India
ABSTRACT
KEYWORDS: Brugia malayi; Heat shock protein 60; Leishmania donovani; Th1 cytokines; Th2 cytokines
1. Introduction
Visceral leishmaniasis (VL) is a vector-borne debilitating neglected tropical disease caused by an obligate protozoan parasite Leishmania donovani (L. donovani). It is a second disease after malaria in the number of fatalities. Including 98 countries of the World, this disease is endemic in tropics, subtropics, and Mediterranean basin regions. In 2015, WHO has included VL in its most neglected tropical diseases list, which immensely affects the liver and spleen in humans. People with extreme poverty, poor hygienic conditions,and living in the endemic regions are more prone to this disease.About 616 million people living in endemic areas are at risk for VL[1].Seven countries from where 90% of cases are reported are India,Brazil, Ethiopia, Kenya, South Sudan, Sudan, and Somalia[2]. In India,the disease is endemic in Bihar, Jharkhand, Eastern Uttar Pradesh,West Bengal, and recently some cases have been reported from Gujarat and Kerala (in women and children 0-9 years of age)[3]. There are complications in the treatment of this disease due to the presence of many causative species, its manifestation in different clinical forms, lengthy treatment procedures, non-availability of vaccine,development of resistance, absence of drug specificity, and toxicity.In the past few years, researchers have focused on the development of an effective vaccine using potential protein antigens/recombinant proteins of the parasite (L. donovani) with different adjuvant systems in rodents. However, to date, no potential vaccine candidate is available.
In the case of VL, the Th1/Th2 immune response patterns of the host play a crucial role in immunity against Leishmania.Th1 cytokines [interferon gamma (IFN-γ), interleukin-12 (IL-12), and tumor necrosis factor-alpha (TNF-α)]produced during cellular immunity increase the expression and production of nitric oxide (NO), which plays a decisive role in parasite clearance and development of protection against the parasite by oxidative burst in infected phagocytes[4,5]. IL-4 and IL-10 cytokines produced from regulatory Th2 cells induce progression of the disease.These Th2 cytokines hampered the protection against infection by regulating the production of IFN-γ from Th1 cells[6,7]. Th17 cells produce prototypic cytokine IL-17 and induce the expression of the transcription factors [related orphan receptor (RORγT); signal transducer and activator of transcription 3 (STAT3)]that act as a protector for VL[8].
The immunological effect of filarial and leishmanial coinfection on immune responses has been primarily demonstrated in the mouse model. Filarial parasite Litomosoides sigmodontis delayed the progression of Leishmania major (L. major) by facilitating pathological wounds in mice, but the outcomes of this coinfection on parasite burden were obscure[9]. Filarial adult parasite Brugia malayi (B. malayi) fraction (BmAFII) stimulates the production of proinflammatory cytokines (IFN-γ, TNF-α, IL-1β, IL-8), NO release, and cellular proliferative responses. These factors might be responsible for inhibiting the progression of L. donovani infection in immunized mice[10]. Furthermore, F6 fraction (54.2-67.8 kDa) of BmAFII fraction inhibits L. donovani infection (~72%)by the upregulation of Th1 cytokines (IFN-γ, TNF-α, IL-12),lymphocyte proliferation, IgG, IgG2/3 levels, NO production and downregulation of Th2 cytokine (TGF-β). Earlier reports revealed that antigenic regions of B- and T-cell epitopes in HSP60 of F6 of B. malayi are common with L. donovani counterparts[11]. On this basis, we have predicted that B. malayi HSP60 (BmHSP60) may have immunostimulatory potential, which inhibits the leishmanial infection. Therefore, in light of these recent studies, the present study was aimed to evaluate the immunostimulatory potential of BmHSP60 using murine macrophage cells (J774A.1), which was characterized by the previous proteomic search. Recombinant B.malayi HSP60 (rBmHSP60) was expressed and purified by the method described by Misra et al. with some modifications[12]. Then the in vitro potency of rBmHSP60 protein as an immune stimulator against VL was evaluated.
2. Materials and methods
2.1. Cell line
Murine macrophage cell line J774A.1 was purchased from National Center for Cell Science, Pune, India. Cells were aseptically maintained in our laboratory in complete RPMI-1640 medium[10% fetal bovine serum (Gibco, USA); penicillin antibiotic: 100 U/mL, streptomycin antibiotic: 100 µg/mL]. For experiments, cell suspension (2 × 10cells/mL) was prepared in complete RPMI-1640 medium, and cell viability was checked by exclusion method with 0.1% trypan blue dye.
2.2. Cloning, expression, and purification of BmHSP60
Live adult parasites of B. malayi were procured from CSIR-CDRI,Lucknow (India). RNA was extracted from adult B. malayi using Trizol method[13]and RevertAidFirst-Strand cDNA Synthesis Kit(Thermo, USA) was used for cDNA synthesis. Primers of BmHSP60 were designed by IDT software. A forward primer was flanked with BamHⅠ restriction site while the reverse primer was flanked with XhoⅠ restriction site. The primers used for cloning were: Forward:5' GGATCCAATGTTCCGTATCGGTGGAC 3'; Reverse: 5'CTCGAGGTACATTCCTCCCATACCTC 3'.
Briefly, ~ 1.7 kb amplicon having complete open reading frame of BmHSP60 gene was amplified by PCR using BmHSP60 specific designed primers and cloned into the pTriEx-4 expression vector. The recombinant vector pTriEx-4-BmHSP60 was transformed in BL21-DE3 competent Escherichia coli strain. Protein was expressed in BL21-DE3 cells with 1 mM isopropyl β-D thiogalactoside (250 µL/250 mL culture, stock concentration: 1 M; pH 7.2) for 24 h at 22 ℃ and cells were harvested by centrifugation at 4 500 rpm for 20 min. The pellet was sonicated in lysis buffer [50 mM Tris pH 8.0, 250 mM NaCl, 10 mM imidazole, Triton-X, 1 000 µM protease inhibitor phenylmethylsulfonyl fluoride (Sigma, USA), 1 mg/mL lysozyme(Sigma, USA)]under chilled conditions[12].
The suspension was centrifuged at 4 500 rpm for 30 min at 4 ℃, and 10 µL of suspension was checked on 10% SDS-PAGE. The suspension was applied to Ni-NTA sepharose column and washed with buffer containing graded concentrations of imidazole (10, 20, 50, and 100 mM) in 50 mM NaHPO, 300 mM NaCl (pH 7.8). rBmHSP60 was eluted using 300 mM imidazole, desalted by dialysis (cut off: 10 kDa), concentrated using Amicon ultrafilters (Millipore, USA) with 10 kDa cut off and finally checked on 10% SDS-PAGE. Bacterial lipopolysaccharide (LPS) was removed by Triton-X method, and purified rBmHSP60 was tested for the level of endotoxin by Limulus amebocyte lysate test (Lonza, USA)[14]. The Bradford assay was performed for quantifying the protein content[15].
2.3. Chaperone assay
Chaperones are a group of proteins that are involved in protein folding[16], protein refolding, translocation of protein through membranes[17], and also facilitate protein degradation[18].They protect cellular proteins from heat damage by thermal aggregation phenomena. The chaperon assay was performed for the determination of the thermal aggregation property of BmHSP60 using citrulline synthase (CS). CS is a natural cellular protein having high sensitivity towards heat denaturation. Thermal aggregation assay was accomplished as described by Gnanasekar et al.[19].
Briefly, different concentrations (1 and 2 µM) of rBmHSP60 were prepared in a buffer containing 50 mM sodium phosphate and 100 mM NaCl (pH 7.4). CS (1 µM) was incubated with different concentrations of rBmHSP60 at a molar ratio of 1:1, 1:2 for different time intervals (0 to 40 min) at 45 ℃. Bovine serum albumin(BSA) was used as a control. Absorbance was taken at 300 nm spectrophotometrically.
2.4. Cell proliferation assay
Cell proliferation was accomplished using 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay as an alternative to thymidine based lymphocyte transformation test.For the assay, macrophage cell line J774A.1 was seeded in 96-well cell culture plates (Nunc, Denmark) at a concentration of 2 ×10cells/well/200 µL prepared in complete RPMI-1640 medium.Cells were stimulated with rBmHSP60 (0.5, 1, 2, 3, and 4 µg/mL)or Concanavalin A (10 µg/mL) and incubated for 72 h at 37 ℃with 5% CO. After 72 h, the medium was removed, and 50 µL of MTT solution (concentration: 0.5 mg/mL) was added per well and incubated for 3 h. Further, the MTT solution was removed, and 100µL of dissolving solution dimethyl sulfoxide (DMSO) was added for dissolution of purple-colored formazan crystals. Absorbance was measured at 570 nm on an ELISA reader (Biotek, USA)[20].
2.5. NO release
NO released from macrophage cells J774A.1 was measured using the Griess reagent. Briefly, cells were dispensed into 48 well (1 × 10cells/500 µL/well) sterile cell culture plates (Genetix, USA) followed by incubation overnight (O/N) at 37 ℃ with 5% CO. After overnight incubation, medium along with non-adherent cells was removed, and fresh medium (500 µL) was added. Further, cells were stimulated with rBmHSP60 (0.25, 0.5, 1, 2, 3, 4 and 5 µg/mL) or LPS (1 µg/mL) for 48 h at the same atmosphere. Production of nitrite in the medium was quantified using the method described by Kushwaha et al.[21].
2.6. Determination of inducible nitric oxide synthase (iNOS),STATs and cytokine expression by qRT-PCR
J774A.1 cells were dispensed into six-well (1 × 10cells/500µL/well) sterile cell culture plates (Genetix, USA) followed by incubation overnight (O/N) at 37 ℃ with 5% CO. After 24 h, cells were replenished with fresh complete medium and stimulated with rBmHSP60 or LPS (1 µg/mL). After 48 h of incubation,stimulated cells were used for gene expression [iNOS, STATs, and cytokine expression (IFN-γ
, TNF-α
, IL-12, IL-6, and IL-10)], and supernatants were assessed for cytokine release.Total RNA was isolated from cells using the Trizol method,and cDNA was prepared using RevertAidFirst-Strand cDNA Synthesis Kit (Thermo, USA) as per the manufacturer's protocol.RT-PCR was carried out in LightCycler480 system (Roche,UK) for analysis of iNOS, various transcription factors (STAT1,STAT3, STAT4), Th1 (IFN-γ
, TNF-α
, IL-12) and Th2 (IL-6, IL-10) cytokines expression[22,11]. The following reaction conditions were performed using IDT software designed primers (Table 1):initial denaturation (95 ℃ for 2 min) followed by 40 cycles, each consisting of denaturation (95 ℃ for 30 s), annealing (55 ℃ for 40 s), and extension (72 ℃ for 40 s per cycle). Unstimulated cells(J774A.1 cells) were used as control for calculating gene expression corresponding to stimulated cells. Fold change in mRNA expression was calculated by using the comparative Cmethod[23].2.7. Cytokine release
Th1 (IFN-γ, TNF-α, IL-12) and Th2 (IL-10) cytokines released from rBmHSP60 stimulated cell culture supernatants for 48 h were measured by ELISA kit (Diaclone, France), according to the manufacturer's guidelines. All samples were run in triplicates. The absorbance of the colored complex was taken at 550 nm.
2.8. Statistical analysis
Statistical analysis of data was performed using GraphPad Prism 6.01 (GraphPad Software, San Diego, USA) using Student's t-test and Tukey's multiple comparison test. Results were presented as mean ± SD of two experiments. P-value < 0.05 was considered significant.
3. Results
3.1. Cloning, expression, and purification of rBmHSP60
The amplified product of BmHSP60 (~1.725 kb) was cloned in TA vector, and the product was verified by colony PCR using BmHSP60 specific primers (Figure 1A and B). The final recombinant constructpTriEx-4-BmHSP60 was confirmed through double digestion of construct by BamHⅠand XhoⅠrestriction enzymes and colony PCR using BmHSP60 specific primer (Figure 1C and 1D). Isopropyl β-D-thiogalactoside induced recombinant protein and Ni-NTA sepharose column purified recombinant HSP60 (~65 kDa) were visualized on 10% SDS-PAGE gel (Figure 2A and 2B). Measurement of endotoxin levels by E-toxateshowed an insignificant endotoxin level (<1 EU/mg).

Table 1. Primer sequences (obtained from NCBI) used in the study.
3.2. Chaperone property of rBmHSP60
Chaperone function is one of the special features of heat shock proteins. A thermal agglutination reaction was performed to analyze the chaperone property of HSP60. Incubation of CS with rBmHSP60(at a molar ratio of 1:1, 1:2) significantly increased the thermal aggregation of CS protein while incubation of CS alone or with BSA(control) remain unfolded at 45 ℃ (Figure 2C).
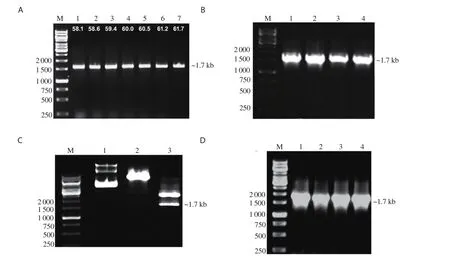
Figure 1. Cloning of BmHSP60. (A) Amplification of Brugia malayi HSP60 gene (BmHSP60) by PCR using HSP60 gene-specific primers, M: 1 kb DNA ladder (Fermentas, USA), Lane 1-7: PCR product (~1.7 kb) of BmHSP60 gene. (B) Colony PCR of transformed white colonies containing BmHSP60 gene in pTZ57R/T (TA cloning vector) using HSP60 specific primers, M: 1 kb DNA ladder, Lane 1-4: PCR product of gene from different colonies, ~1.7 kb PCR product shows successfully cloned BmHSP60 gene in TA vector. (C) Double digestion of plasmid of pTZ57R/T-BmHSP60 gene construct with BamHⅠ and XhoⅠ restriction enzymes, M: 1 kb DNA ladder, Lane 1: Undigested pTZ57R/T, Lane 2: Digested pTZ57R/T, Lane 3: Digested pTZ57R/T-BmHSP60 gene construct, respectively. (D) Colony PCR of BmHSP60 gene cloned in pTriEx-4 vector using HSP60 gene-specific primers, M: 1 kb DNA Ladder (Fermentas,USA), Lane 1-4: PCR product of BmHSP60 gene from different colonies, ~1.7 kb band represents BmHSP60 gene insert.
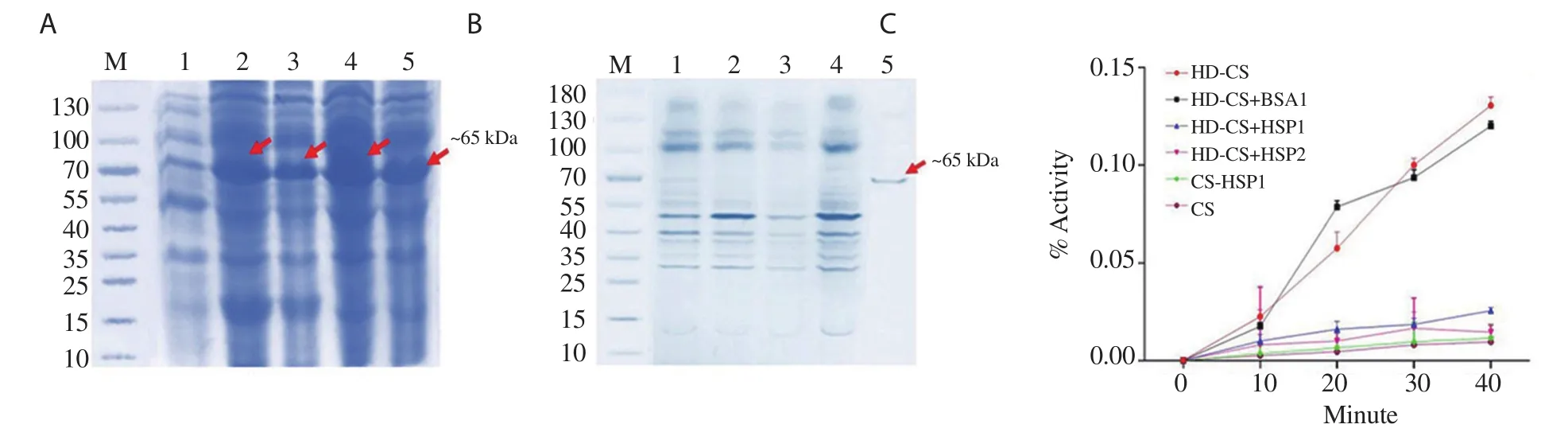
Figure 2. (A) Expression of recombinant BmHSP60 (rBmHSP60) protein in the cell lysate of transformed Escherichia coli BL21-DE3 with recombinant plasmid and induced with isopropyl β-D-thiogalactoside (IPTG). M: Molecular weight marker (Fermentas, USA), Lane 1: uninduced cells transformed with recombinant plasmid, Lane 2: IPTG induction of 0.5 mM at 37 ℃ for 4 h, Lane 3: IPTG induction of 0.75 mM at 37 ℃ for 4 h, Lane 4: IPTG induction of 1.0 mM at 25 ℃ for 4 h, Lane 5: IPTG induction of 1.5 mM at 25 ℃ for 4 h. (B) Purification of rBmHSP60 by affinity chromatography and 10% SDS PAGE.M: Molecular weight marker (Fermentas, USA), Lane 1-5: Eluted rBmHSP60 at different concentrations (10, 20, 50, 100, 300 mM) of imidazole. A single band corresponding to ~65 kDa at 300 mM represents purified rBmHSP60 protein. (C) Chaperone assay of recombinant BmHSP60. Heat denatured citrulline synthase (HD-CS) was incubated alone, in the presence of 1 µM of bovine serum albumin (BSA), 1 µM of BmHSP60, or 2 µM of BmHSP60 while 1 µM of CS was incubated with 1 µM of BmHSP60. CS was incubated without BmHSP60.
3.3. rBmHSP60 promotes cell proliferation and NO release
The proliferative response of J774A.1 macrophage cells was assessed by MTT assay. rBmHSP60 induced significantly higher proliferation of J774A.1 cells as compared to unstimulated cells (P < 0.001, Figure 3A). Moreover, NO production was significantly enhanced when stimulated with rBmHSP60 in vitro (P < 0.001, Figure 3B).
3.4. Expression of STATs and iNOS in J774A.1 macrophages
Higher expression of STAT1 and STAT4 was found in rBmHSP60 stimulated cells while there was no change in STAT3 expression in response to rBmHSP60 (Figure 4A and C). The iNOS level was upregulated by ~1.5 fold (P < 0.05, Figure 4A and C) in the rBmHSP60 stimulated cells as compared to unstimulated cells.
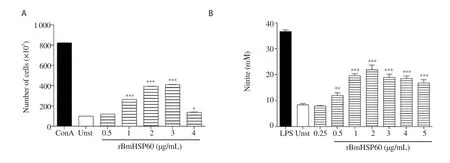
Figure 3. (A) Proliferative activity of J774A.1 cells in response to rBmHSP60 (0.5-4 µg/mL) or Con A (10 µg/mL) in vitro. (B) Nitric oxide released by J774A.1 cells in response to rBmHSP60 (0.25-5 µg/mL) or LPS (1 µg/mL) in vitro. The values are expressed as mean ± SD of data in two experiments.Student's t test was used for statistical analysis. *P<0.05, **P<0.01, ***P<0.001 (vs. unstimulated). Unst: Unstimulated, Con A: concanavalin A, LPS:lipopolysaccharide.
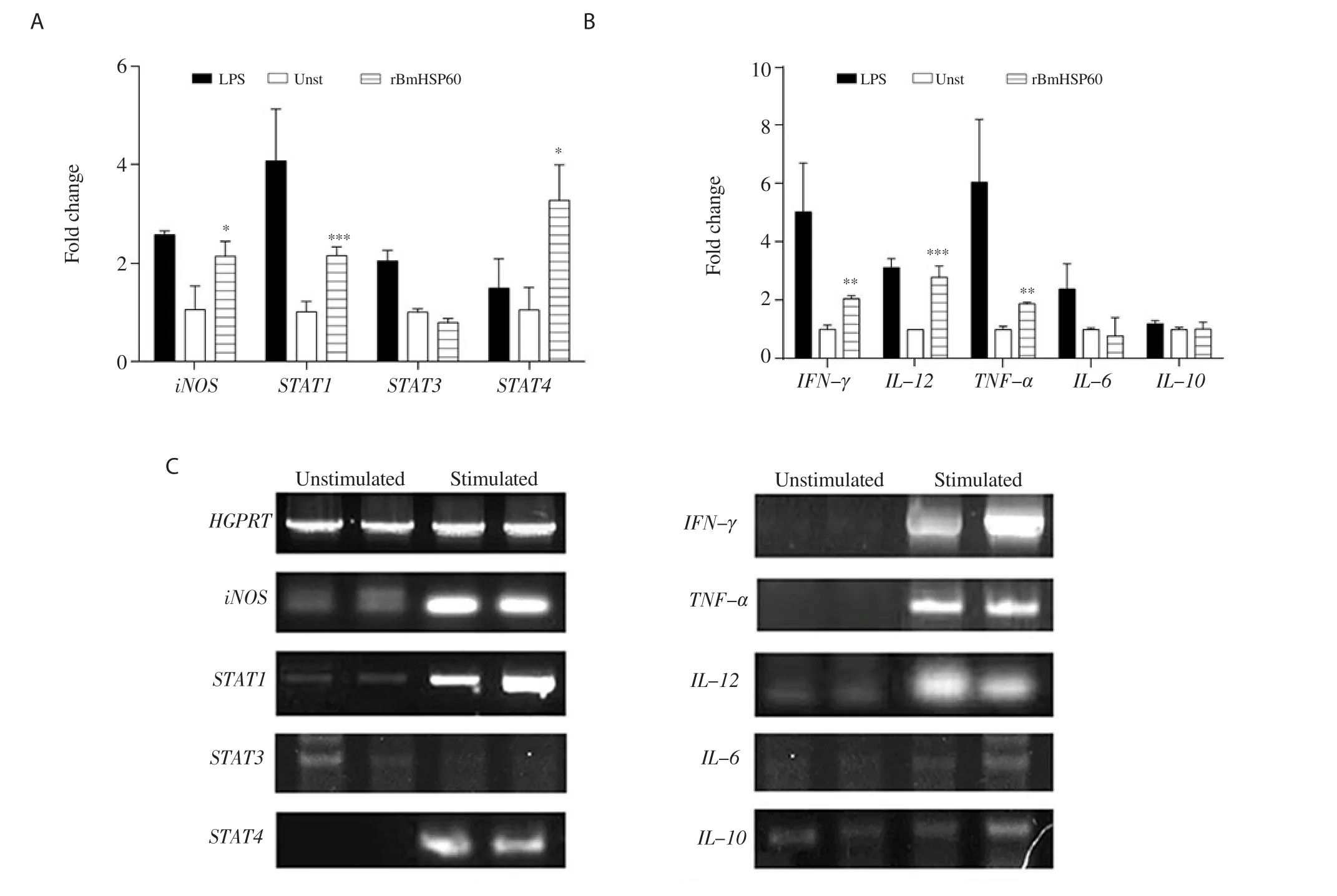
Figure 4. Expression of iNOS, STAT1, STAT3, STAT4, IFN-γ, TNF-α, IL-12, IL-6, and IL-10 cytokine mRNAs by quantitative real-time RT-PCR in macrophage cell line J774A.1 on stimulation with rBmHSP60 (1 µg/mL) in vitro. (A-B) Bar graphs show the quantitative results of mRNA expression. (C)Representative bands. HGPRT was used as a housekeeping control in qRT-PCR results. The values are expressed as mean ± SD of data in two experiments.Student's t test was used for statistical analysis. *P<0.05, **P<0.01, ***P<0.001 (vs. unstimulated).
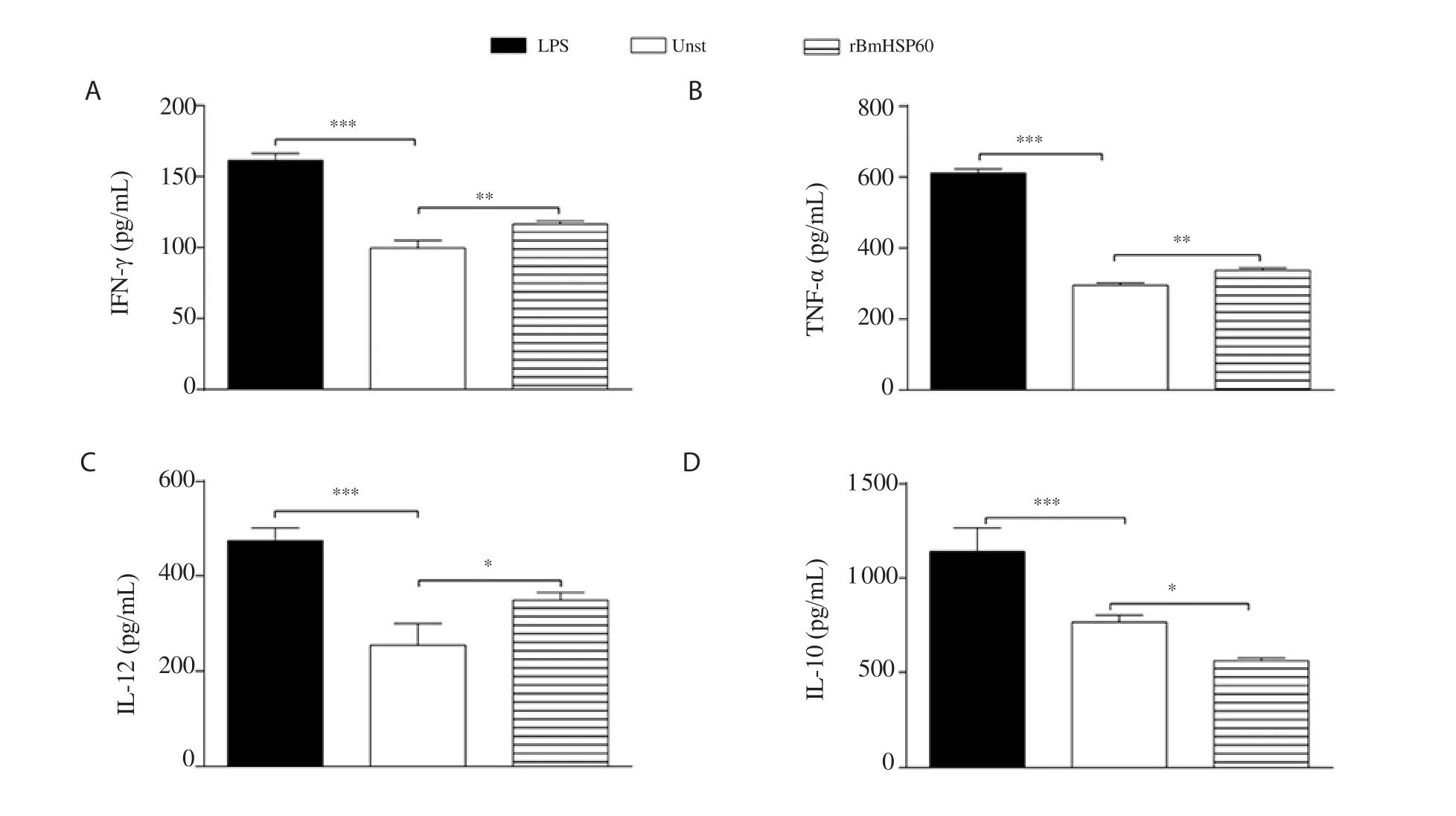
Figure 5. Cytokine levels (A: IFN-γ, B: TNF-α, C: IL-12, and D: IL-10) of macrophage cell J774A.1 in response to rBmHSP60 (1 µg/mL) or LPS (1 µg/mL)in vitro. The values are expressed as mean ± SD of data in two experiments. Student's t test was used for statistical analysis. *P<0.05, **P<0.01, ***P<0.001 (vs.unstimulated).
3.5. Expression and release of cytokines in J774A.1 macrophages
The expression of Th1 cytokines including IFN-γ
, TNF-α
, and IL-12 was found to be ~1.5-2.0 fold higher in stimulated cells in comparison to unstimulated cells (Figure 4B and C). In contrast,no change was observed in the expression of Th2 cytokines (IL-6 and IL-10) (Figure 4B and C). Stimulation of macrophages with rBmHSP60 increased the IFN-γ (P < 0.01; Figure 5A), TNF-α (P <0.01; Figure 5B), and IL-12 (P < 0.05; Figure 5C) cytokine release in vitro but not IL-10 (Figure 5D) as compared to unstimulated cells.4. Discussion
The development of an improved, safer, and better-defined effective vaccine against leishmaniasis, is urgently required. Now, vaccine development against most diseases has shifted towards the discovery of a subunit vaccine, which would be better and safer. Thus, there is a need for a potent antigen molecule having antileishmanial properties with immunostimulatory potentials. HSPs are potential immunostimulatory molecules that act as a bridge between innate and adaptive subsets of the immune system[24]. During the generation of cell-mediated immunity, HSP-chaperoned peptides bind to surface receptors (MHC-I) of macrophages and dendritic cells. These peptides are further presented to T lymphocytes to activate them[25].Various HSP molecules, like Hsp60, Hsp70, Hsp90, and gp96 activate macrophages, lymphocytes, dendritic cells (APCs) and have been reported to be potent activators of the innate immune system[26].HSPs induce the production of proinflammatory cytokines that participate in the pathogenesis of chronic inflammatory and autoimmune diseases. Initially, it was predicted that HSPs stimulate IL-1 or TNF-α cytokine production from monocytes/macrophages or cell lines. Later on, other studies suggested that Hsp60 stimulates the release of cytokines IL-1, TNF-α, IL-6, IL-12, IL-15, and NO production from monocytes/macrophages, elevates the expression of CD62e, CD106, CD54 from endothelial cells, and IL-6 from vascular smooth muscles[27]. Due to the great similarity between parasite and human HSPs, parasite originated HSPs are chief immunogens to humans[28]. The amino acid sequence of human and Trypanosoma cruzi HSP70 showed ~73% similarity, which makes Trypanosoma cruzi HSP70 as a strong immunogen against Chagas disease[29]. Similarly, B- and T-cell epitopes of HSP60 of B. malayi, and L. donovani are common, which may induce strong immune responses against these parasitic diseases[11]. Based on these findings, to assess the immunostimulatory potential of crossreactive molecule HSP60, we cloned HSP60 protein of B. malayi in expression vector pTriEx-4, and this protein was used to stimulate cells of murine macrophages cell line J774A.1. The findings of this study demonstrated that this protein enhanced the mRNA expression of STATs, Th1 cytokines (IFN-γ
, TNF-α
, IL-12), iNOS with increased production of NO and Th1 cytokine levels (IFN-γ, TNF-α,IL-12). These preliminary observations in macrophages suggested the immunogenic nature of filarial and Leishmania cross-reactive molecule HSP60.In this present study, the proliferative efficacy of macrophage cells after treatment with rBmHSP60 was analyzed by MTT assay. The colorimetric MTT assay was used to assess the proliferation and survival of cells[30,20]. MTT is a non-radioisotopic,colorimetric method in which dehydrogenase enzymes released from metabolically active cells reduce the tetrazolium salts into formazan product (purple colored). This method is used in studies for chemotherapeutic drug testing, detection and quantitation of cytokines, immune-cytotoxic activity, chemo- and radio-sensitivity,evaluation of filarial parasite viability, measurement of peripheral blood lymphocytes proliferation and cell death[31,32]. The results illustrated that cells treated with different concentrations of rBmHSP60 (0.5-4 µg/mL) showed increased cell proliferation initially, which decreased after an increase in its concentration. We concluded that 1 µg/mL of rBmHSP60 caused a significant increase in cell proliferation in vitro as compared to unstimulated cells.This result suggested that rBmHSP60 is a potent stimulator of cell proliferation and may be implicated in protection against Leishmania infection.
We also demonstrated the effect of rBmHSP60 on NO production by murine macrophage J774A.1 cells. rBmHSP60 induced significant NO production by macrophages, which also supported the view regarding the control of parasitic infections, which are mediated by upregulated iNOS and NO-mediated macrophage effector mechanisms. NO is a major defense molecule of immune cells. During leishmanial infection, intracellular parasite killing and control of parasite infections are mediated by the production of several mediators, principally by NO, which is released from activated macrophages after exposure of parasite-specific T cells derived IFN-γ[33,34].
JAK-STAT signaling pathway transduces the signals to nucleus that regulates biological processes like cell proliferation, differentiation,cell migration, and apoptosis[35,36]. During stimulation, STAT factors phosphorylate, dimerize, and translocate to the nucleus, where they activate transcription. STAT activation occurs in response to cytokines. We further assessed whether the stimulation of J774A.1 cells by rBmHSP60 modulates the expression of STATs. Results demonstrated that rBmHSP60 stimulated cells showed significant expression of STAT1 and STAT4 while no change in expression of STAT3. Studies undertaken earlier by researchers have demonstrated that infection by pathogens causes the release of IFN-γ and other cytokines IFN-αβ and IL-27, which activate STAT1 and STAT2.Studies on STAT1 deficient mice showed that STAT1 is a key molecule for the proper function of CD4+ and CD8+ T cells, and its deficiency caused a reduction in IFN-γ production[37]. Due to low levels of IFN-γ, STAT1 deficient mice are susceptible to L. major infection[38]. Butcher and his colleagues reported that when macrophages are infected with Taxoplasma gondii, STAT3 suppressed the production of pro-inflammatory cytokines (IL-6, IL-10), but it is not clear how STAT3 regulates their pathway[39].
Additionally, change in mRNA expression profile and release of Th1 (IFN-γ, TNF-α, IL-12) and Th2 cytokines (IL-6 and IL-10) was assessed in rBmHSP60 stimulated cells. Our results of cell-mediated immune response showed that rBmHSP60 enhanced significantly the expression of iNOS along with IFN-γ
, TNF-α
, IL-12 and release of these cytokines (IFN-γ, TNF-α, IL-12) which demonstrated that rBmHSP60 has immunogenic properties and production of cellmediated responses is predominantly of Th1-type. However, there was no significant difference found in the expression of Th2 (IL-6 and IL-10) cytokines. A higher mRNA expression and release of these cytokines suggest stimulation of T cells and macrophages that might have significant functional implications in the defense against leishmanial infections. IFN-γ performed the killing of intracellular pathogens by activating macrophage phagocytosis[40].During leishmaniasis, IFN-γ induced iNOS and NO production by macrophages that play an important role in clearance of Leishmania infection[38,41]. IFN-γ signal protects the host from L. major infection via a STAT1-mediated pathway. Release of IFN-γ and IL-12 through their signaling pathways depends on the activation of STAT1 and STAT4 transcription factors, which regulate the Th1 and inflammatory responses[38]. During Leishmania infection,suppression of pro-inflammatory cytokine IL-12 facilitates the entry of parasite into host macrophages. Earlier reports also indicated that IL-12 expression induces the IFN-γ production by T and NK cells that might be involved in protection against intracellular pathogens like virus, bacteria, and parasitic infections[42]. TNF-α induces reactive nitrogen intermediates production either alone or with IFN-γ to generate cytotoxic effect against pathogens. TNF-α appears to play an essential role in host resistance and decrease the parasite burden and lesion size in cutaneous leishmaniasis[43].Remarkably higher mRNA expression and release of Th1 cytokines(IFN-γ, TNF-α, IL-12) in stimulated cells suggests that rBmHSP60 has immunostimulatory potential, which might help in the reduction of Leishmania parasites by activation of Th1 helper cells and macrophages.In conclusion, we demonstrate that a cross-reactive molecule of B.malayi and L. donovani rBmHSP60 is immunogenic and effectively triggers the Th1 response of murine macrophages. Results correlate with the enhanced expression of STATs, Th1 cytokines, iNOS with increased production of NO, and Th1 cytokines (IFN-γ, TNF-α, IL-12). This study provides preliminary data for further investigations of the immunoprophylactic efficacy of BmHSP60 in rodent model against experimental VL.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Acknowledgments
The authors express special thanks to Department of Science and Technology, Science and Engineering Research Board, New Delhi,India for providing financial support for this work.
 Asian Pacific Journal of Tropical Biomedicine2021年1期
Asian Pacific Journal of Tropical Biomedicine2021年1期
- Asian Pacific Journal of Tropical Biomedicine的其它文章
- Pharmacological aspects of fisetin
- Sang-Yod rice bran hydrolysates alleviate hypertension, endothelial dysfunction,vascular remodeling, and oxidative stress in nitric oxide deficient hypertensive rats
- Network pharmacology-based analysis of effective components and mechanism of Rhizoma coptidis in treating diabetes
- Identification of climatic and environmental factors associated with incidence of cutaneous leishmaniasis in Central Iran using satellite imagery