
口腔清洁护理用品 牙膏用甘草次酸(T/COCIA1-2018)
2021-12-09 07:45
口腔护理用品工业 2021年5期
前言
本标准按照GB/T 1.1-2009给出的规则起草。
本标准由中国口腔清洁护理用品工业协会提出。
本标准由全国口腔护理用品标准化技术委员会牙膏分技术委员会(SAC/TC492/SC1)归口。
本标准起草牵头单位:甘肃泛植制药有限公司。
本标准共同制定单位:云南白药集团股份有限公司、柳州两面针股份有限公司、广州薇美姿实业有限公司、重庆登康口腔护理用品股份有限公司、苏州市金茂日用化学品有限公司、江西草珊瑚口腔护理用品有限公司、江苏雪豹日化有限公司。
本标准主要起草人:赵富虎、陈保华、陈国宝、高鹰、黄华来、陈敏珊、蒋玮、陈健芬、许海燕、徐志良。
本标准为首次发布。
1 范围
本标准规定了牙膏用甘草次酸的产品质量要求、试验方法、检验规则及标志、包装、运输、贮存、保质期。
本标准适用于以甘草(豆科植物甘草Glycyrrhiza uralensis Fisch.、胀果甘草Glycyrrhiza inflata Bat.及光果甘草Glycyrrhiza glabra L.)为主要原料,经过提取、乙酰化、碱解、结晶纯化等工艺制成的甘草次酸。本标准同样适用于牙膏、口腔清洁护理液、义齿稳固剂等口腔护理清洁用品的原料。
2 规范性引用文件
下列文件对于本文件的应用是必不可少的。凡是注日期的引用文件,仅所注日期的版本适用于本文件。凡是不注日期的引用文件,其最新版本(包括所有的修改单)适用于本文件。
GB/T 191 包装储运图示标志
GB/T 601 化学试剂 标准滴定溶液的制备
GB/T 602 化学试剂 杂质测定用标准溶液的制备
GB/T 603 化学试剂 实验方法中所用制剂及制品的制备
GB/T 6678 化工产品采样总则
GB/T 6682 分析实验室用水规格和实验方法
GB/T 8170 数值修约规则
JJF 1070 定量包装商品净含量计量检验规则
QB/T 4821 口腔清洁护理用品 牙膏中汞含量的测定方法
QB/T 4822 口腔清洁护理用品 牙膏中砷含量的测定方法
《定量包装商品计量监督管理办法》(中华人民共和国质量监督检验检疫总局令[2005]第75号)
《中华人民共和国药典》2015版(四部)
《化妆品安全技术规范》2015版
3 技术要求
3.1 感官要求、理化指标
应符合表1的规定。
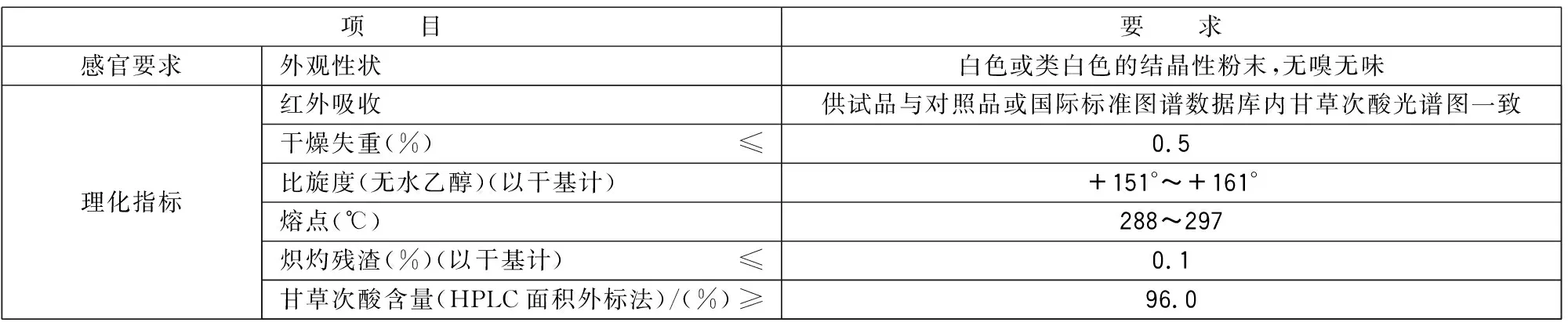
表1 感官要求、理化指标
3.2 杂质、卫生要求
应符合表2规定。

表2 杂质、卫生要求
3.3 净含量
包装产品的净含量应符合《定量包装商品计量监督管理办法》(中华人民共和国国家质量监督检验检疫总局令〔2005〕第75号)的规定。
4 试验方法
4.1 一般规定
本标准所用试剂和水,在没有注明其他要求时,均指分析纯试剂和GB/T 6682规定的三级水。实验中所用标准溶液、杂质标准溶液、制剂及制品,在没有注明其他要求时,均按GB/T 601、GB/T 602、GB/T 603的规定制备。试验中所用溶液在未注明用何种溶剂配制时,均指水溶液。
原始样品经充分混匀后作为试验样品(简称“试样”)。
4.2 感官要求
取适量试样,置于清洁、干燥的玻璃平皿中,在自然光线下,用目测法判定外观。同时感官检查其滋味及气体。
4.3 理化指标
4.3.1 红外吸收
4.3.1.1 仪器
傅里叶变换红外光谱仪或色散型分光光度计。
4.3.1.2 试剂
溴化钾(光谱纯)。
4.3.1.3 测试方法
取样品0.01g~0.05g,按100~200倍取溴化钾,共同研磨,取研磨好的混合物进行压片测试。对照品相同方法处理。
4.3.1.4 结果判定
检查供试品红外吸收光谱图与对照品或国际标准图谱数据库内甘草次酸光谱图是否一致。
4.3.2 干燥失重
4.3.2.1 仪器
电热恒温干燥箱。
分析天平:感量0.1mg。
4.3.2.2 操作方法
取约1g试样,置于105℃干燥至恒重的扁形称量瓶中,精密称定。
在105℃干燥2h。干燥时,应平铺在扁形称量瓶中,厚度不可超过5mm。放入烘箱或干燥器进行干燥时,应将瓶盖取下,置称量瓶旁,或将瓶盖半开进行干燥;取出时,应将称量瓶盖好。置烘箱内干燥的试样,应在干燥后取出置于干燥器中放冷至室温,然后称定其质量。
根据减失的质量和取样量计算试样的干燥失重。
4.3.2.3 结果计算
干燥失重L1按公式(1)计算:
(1)
式中:m2——干燥前试样加称量瓶的质量,单位为克(g);
m3——干燥后试样加称量瓶的质量,单位为克(g);
m1——试样质量,单位为克(g)。
试验结果以平行测定结果的算术平均值为准(保留1位小数)。在重复性条件下获得的两次独立测定结果的绝对差值不应大于1.0%。
4.3.3 比旋度
4.3.3.1 试剂和材料
无水乙醇。
4.3.3.2 仪器和设备
旋光光度计,最小分度值0.05°。
4.3.3.3 操作方法
取试样1.0g,用无水乙醇溶解并稀释至100mL测定旋光度。
4.3.3.4 结果计算
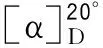
(2)
式中:α——试样溶液测得的旋光度,单位为度(°);
100——溶解试样的溶液体积,单位为毫升(mL);
l——旋光管长度,单位为分米(dm);
m1——试样质量,单位为克(g);
w1——试样的干燥失量,%。
取3次平行测定结果的算术平均值为测定结果,保留至整数位。
4.3.4 熔点
按照《中华人民共和国药典》2015年版四部,0612熔点测定项下第一法中的毛细管熔点法测定。
4.3.5 炽灼残渣
4.3.5.1 仪器和设备
高温炉。
分析天平:感量0.1mg。
4.3.5.2 操作方法
取试样约1g精确至0.0001g,干燥后的样品,精密称定,缓缓炽灼至完全炭化,放冷;加硫酸0.5mL~1mL使湿润,低温加热至硫酸蒸气除尽后,在700℃~800℃炽灼使完全灰化,移置干燥器内,放冷,精密称定后,再在700℃~800℃炽灼至恒重,即得。若需将残渣留作重金属检查,则炽灼温度应控制在500℃~600℃。
注:除另有规定外,恒重系指试样连续两次炽灼后称重的差异在0.3mg以下的情况。炽灼至恒重的第二次称重应在继续炽灼30min后进行。
4.3.5.3 结果计算
炽灼残渣以质量分数w1按公式(3)计算:
(3)
式中:m4——炽灼后坩埚加灰分的质量,单位为克(g);
m5——坩埚的质量,单位为克(g);
m1——试样质量,单位为克(g)。
试验结果以平行测定结果的算术平均值为准(保留1位小数)。在重复性条件下获得的两次独立测定结果的绝对差值不应大于1.0%。
4.3.6 甘草次酸含量
4.3.6.1 仪器
高效液相色谱仪:配有紫外检测器或光电二极管阵列检测器。
天平:感量0.01mg。
电热恒温干燥箱。
4.3.6.2 试剂和材料
乙腈:色谱纯。
冰乙酸:优级纯。
0.45μm滤膜(有机膜)。
甘草次酸对照品(国家药品标准物质-中国食品药品检定研究院)。
4.3.6.3 色谱条件
色谱柱: 十八烷基硅烷键合硅胶为填充剂(4.6mm.×250mm,粒径5μm)。
流动相:乙腈、0.2mol/L醋酸铵、冰乙酸的体积比为780.0∶160.8∶79.2。
检测波长: 254nm。
柱温:35℃。
进样量:20μL。
流量:1.000mL/min。
4.3.6.4 对照品溶液的制备
取试样约20mg (精确至0.1mg),精密称定,置于100mL量瓶中,加流动相溶解并稀释至刻度,摇匀,即得。
4.3.6.5 供试品溶液的制备
取试样约20mg (精确至0.1mg),精密称定,置于100mL量瓶中,加流动相溶解并稀释至刻度,摇匀,滤过,弃去初滤液,取续滤液作为供试品溶液。
4.3.6.6 操作方法
精密量取对照品溶液及供试品溶液各20μL,分别注入液相色谱仪,根据各自的峰面积,按公式(4)计算甘草次酸含量w3:
(4)
式中:At——供试品溶液中甘草次酸的峰面积;
ms——对照品溶液中试样称取量,单位为毫克(mg);
w2——对照品溶液中甘草次酸含量(按干燥品计算),%;
Ls——对照品试样干燥失重,%;
As——对照品溶液中甘草次酸的峰面积;
mt——供试品溶液中试样称取量,单位为毫克(mg);
Lt——供试品试样干燥失重,%。
4.4 杂质指标
4.4.1 相关物质
4. 4.1.1 高效液相色谱面积归一化法
测量各杂质峰的面积和色谱图上除溶剂峰以外的总色谱峰面积,计算各杂质峰面积及其之和最大杂质峰面积,占总峰面积的百分率。
4.4.1.2 仪器、材料、色谱条件和供试品溶液的制备
仪器、材料、色谱条件和供试品溶液的制备同4.3.6的相关规定。
4.4.1.3 计算方法
扣除溶剂峰后,采用面积归一化法, 计算最大杂质和各杂质峰面积总和,并计算占总峰面积的百分率。
4. 4.2 溶液的澄清度
取试样0.2g加乙醇30mL使其溶解,观察溶液。
4.4.3 色调
取试样1.25g,精密称定,置于25mL容量瓶中,加无水乙醇溶解并稀释至刻度,在400nm波长处测定吸光度(以无水乙醇作对照)。
4.4.4 残留乙醇
按照《中华人民共和国药典》2015年版四部0861残留溶剂测定法第二法(气相色谱法)测定。
4.4.5 氯化物
4.4.5.1 仪器和试剂
纳氏比色管、标准氯化钠溶液、硝酸银试液、稀硝酸。
4.4.5.2 标准氯化钠溶液的制备
称取氯化钠0.165g,置于1000mL量瓶中,加水适量使溶解并稀释至刻度,摇匀,作为贮备液。临用前,精密量取贮备液10mL,置于100mL量瓶中,加水稀释至刻度,摇匀,即得(每1mL相当于10μg的Cl-)。
4.4.5.3 硝酸银试液的制备
取硝酸银17.5g,加水适量溶解至1000mL,摇匀。置于具有玻璃塞的棕色试剂瓶中,避光保存。
4.4.5.4 稀硝酸的制备
取硝酸(HNO3)10.5mL,加水稀释至100mL,即得。本液含HNO3应为9.5%~10.5% (质量分数)。
4.4.5.5 操作方法
取试样0.1g,置于纳氏比色管中,加乙醇30mL溶解,加水4mL;再加稀硝酸10mL及硝酸银试液1mL,加水稀释使成50mL,摇匀。在暗处放置5min后, 即得供试溶液。供试溶液若带颜色,除另有规定外,可取供试溶液两份,分置50mL纳氏比色管中,一份加入硝酸银试液1.0mL,摇匀,放置10min,若显浑浊,可反复过滤,至溶液完全澄清,再加标准氯化钠溶液5.0mL与水使成50mL,在暗处放置5min,作为对照溶液;另一份加入硝酸银试液1.0mL与水使成50mL,摇匀,在暗处放置5min;与对照溶液同置于黑色背景上,从比色管上方向下观察、比较。
4.4.6 硫酸盐
4.4.6.1 试剂及仪器
纳氏比色管(50mL) ; 25%氯化钡溶液、稀盐酸、标准硫酸钾溶液。
4.4.6.2 标准硫酸钾溶液制备
称取硫酸钾0.181g,置于1000mL量瓶中,加水适量使溶解并稀释至刻度,摇匀,即得(每1mL相当于100μg的SO42-)。
4.4.6.3 氯化钡溶液(25%)制备
取氯化钡25g,加水溶解使成100mL,即得。
4.4.6.4 稀盐酸制备
取盐酸234mL,加水稀释至1000mL,即得。
4.4.6.5 操作方法
取试样0.2g,置于纳氏比色管中,加乙醇30mL溶解,加水4mL;再加稀盐酸2mL及25%氯化钡溶液5mL,加水稀释使成50mL,摇匀;取标准硫酸钾溶液3mL,置于50mL纳氏比色管中,加水使成约40mL,加稀盐酸2mL,摇匀,即得对照溶液。于供试溶液与对照溶液中,分别加入25%氯化钡溶液5mL,用水稀释至50mL,充分摇匀,放置10min,同置于黑色背景上,从比色管上方向下观察与对照液比较。
4.5 有害物质
4.5.1 铅
按照《化妆品安全技术规范》2015年版铅的测定 第一法 石墨炉法原子吸收光谱法进行测定。
4.5.2 镉
按照《化妆品安全技术规范》2015年版镉的测定 火焰原子吸收分光光度法进行测定。
4.5.3 砷
按照QB/T 4822测定。
4.5.4 汞
按照QB/T 4821测定。
4.6 卫生指标
在无菌环境下,称取试样10g加至盛有90mL无菌磷酸盐缓冲液的稀释瓶中,充分振荡混匀,用其作为1∶10的样品稀释液,其余操作程序按《化妆品安全技术规范》2015年版中规定的方法进行检验。
4.7 净含量
按JJF1070中规定的方法进行检验。
5 检验规则
5.1 检验分类
5.1.1 出厂检验
本标准技术要求中的性状、澄清度、色调、干燥失重、炽灼残渣、红外图谱、熔点、比旋光度、甘草次酸含量、杂质限量、菌落总数、霉菌和酵母菌总数、耐热大肠菌群为出厂检验项目。
5.1.2 型式检验
本标准技术要求中所规定的全部项目为型式检验项目,正常情况下每3个月进行1次型式检验。出现以下情况也应进行型式检验:
(a)更新关键生产工艺时;
(b)主要原料有变化时;
(c)停产6个月以上又恢复生产时;
(d)国家质量监督机构或用户提出进行型式检验要求时;
(e)相应法律法规发生改变时。
5.1.3 接收检验
顾客有权按照供需双方所签订合同的有关条款及本标准的规定,对所收到的牙膏用甘草次酸进行验收,验收应在货到之日起的15日内完成。
5.2 组批与抽样规则
5.2.1 组批
5.2.1.1 以一次交货的同规格产品为一批。
5.2.1.2 产品应先由生产企业质检部门按本标准规定检验合格,出具检验报告方可出厂,收货单位按本标准验收。
5.2.2 抽样
5.2.2.1 每个检验批的产量不应超过生产厂每班的产量。据物料单元数目按GB/T 6678的规定确定采样单位数,选定采样单位后,打开包装一次采取微生物检验用样及其他检验用样。
5.2.2.2 采样:微生物检验用样的采样按无菌操作要求进行;其他检验用样将采样器插入采样单元料层的3/4处采取样品,取出不少于100g的样品,采出的样品迅速混匀,经缩分后分装于清洁、干燥的容器中,微生物检验用样的制样按无菌操作要求进行。样品容器应粘贴标签,注明:生产厂名、产品名称、生产批号、采样日期和采样者姓名。样品一份用于检验,另一份保存3个月备查。待检样品应置室内常温、干燥密闭保存。
5.3 判定规则
检验结果按照GB/T 8170修约至规定位数,对照限定值确定检验的产品是否可通过验收。检验结果若不符合要求,应重新自两倍的包装中采样复验,复验结果仍不符合要求时,则整批产品作不合格品处理。
5.4 仲裁
若交收双方对产品检验结果发生异议,双方可协商解决,必要时,可共同选定仲裁机构按本标准检验裁定。
5.5 存样和样本保留
每批验收产品需保留样品作存样,样本保留至保质期满后半年。
6 标志、包装、运输、贮存、保质期
6.1 标志
包装容器上都应贴有牢固明显的标志,内容包括:产品名称、生产厂名、厂址、商标、生产批号或生产日期、净重等。
6.2 包装
本产品要求用内有双层医药级聚乙烯塑料袋,外包装用瓦楞纸箱或纸板桶,也可与用户协商确定。
6.3 运输
本产品在装卸、运输过程中应放在清洁、干燥的场所,不应与有毒物混装、混运或一起存放。
6.4 贮存
本品应避光、密封贮存
6.5 保质期
在符合本标准包装、运输和贮存条件下,自生产之日起保质期为5年,超过保质期可重新检验,检测结果符合本标准要求时,产品仍可使用。
猜你喜欢
草业科学(2022年9期)2022-10-21
食品工业科技(2022年20期)2022-10-11
一重技术(2022年2期)2022-05-12
河南农业·综合版(2022年2期)2022-03-18
河南农业(2022年2期)2022-03-14
河南农业·综合版(2021年7期)2021-08-23
家庭百事通·健康一点通(2020年8期)2020-09-08
爆炸与冲击(2020年8期)2020-08-26
中学化学(2016年12期)2017-02-05
