
连圈状秕糠疹一例
2021-12-08 03:53吴亚光宋志强翟志芳
实用皮肤病学杂志 2021年5期
汪 婷,吴亚光,张 恋,宋志强,翟志芳
临床资料
患者,男,51岁。因躯干、四肢褐色斑片10年余,于2018年3月10日就诊。10余年前,无明显诱因患者腹部出现一处掌心大小褐色斑疹,表面细小鳞屑,无自觉症状,皮损逐渐增多扩大,延至躯干及四肢。外用糖皮质激素制剂无明显好转。患者既往体健,2014年曾因胆囊结石行手术治疗,否认其他系统疾病史,否认特殊接触史,家族成员中无类似疾病。体格检查:一般情况良好,各系统查体无明显异常,未触及增大浅表淋巴结。皮肤科情况:躯干及四肢可见大小不等的类椭圆形褐色斑片,界限相对清楚,部分斑片重叠分布,腹部皮损融合成大片状,表面干燥,有糠秕状鳞屑(图1)。皮损真菌涂片检查阴性。实验室及辅助检查均无异常。取左侧腹部皮损行组织病理检查:表皮轻度角化过度,颗粒层消失,棘层变薄。真皮浅层无明显炎性细胞浸润(图2)。诊断:连圈状秕糠疹。治疗:口服阿维A胶囊,外用复方氟米松乳膏。1个月后随访,患者诉皮损无明显好转自行停药。2年后随访,患者诉皮损无明显变化,自诉系统体检无异常发现。
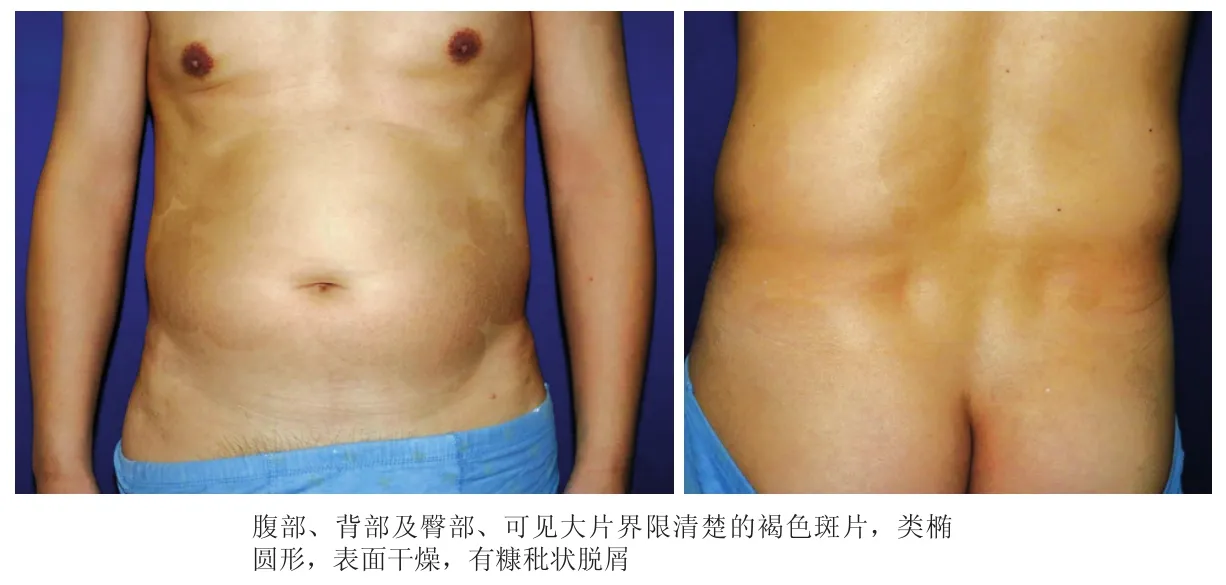
图1 连圈状秕糠疹患者躯干部皮损

图2 连圈状秕糠疹患者腹部皮损组织病理(HE染色)
讨论
连圈状秕糠疹(pityriasis circinata)又称正圆形秕糠疹(pityriasis rotunda)、正圆形后天性假性鱼鳞病(pseudoichthyosis acquisita en taches circulaires),是一种少见的轻度角化过度性皮肤病,1906年由Toyama首次报道。至今本病病因及发病机制仍不明确。有人认为本病是潜在的副肿瘤性皮肤病(如肝细胞癌、IgG骨髓瘤、多发骨髓瘤、白血病、食道癌、胃癌)等,并与多种系统性疾病有关,如营养不良、糖尿病、感染、激素紊乱等[1]。近年来国内外均相继有家族史的报道,根据对家族史的遗传家系分析提示本病存在常染色体显性遗传特征[2,3],且认为其与遗传因素有关,可能是鱼鳞病的一种亚型。另有研究证实患者表皮中缺少丝聚蛋白-2 的表达[4],Hashimoto等[5]采用免疫组化方法检测皮损区中间丝相关蛋白和兜甲蛋白的表达明显减少,提示皮损区角化分化末期的功能障碍与发病密切相关。
Grimalt等[6]将本病分为两型:Ⅰ型见于亚洲人和黑种人,多数表现为色素沉着性损害,皮损数目<30个,认为可能与内在恶性肿瘤和系统性疾病有关,无阳性家族史;Ⅱ型多见于白种人,表现为色素减退性皮损,数量>30个,可有家族性发病,不伴有系统性疾病。但是上述分类并不能包含所有的病例。近年来有关于黑人家族史的报道,中国也相继报道过有阳性家族史的病例。本病组织病理学上表皮的改变类似寻常型鱼鳞病,表现为轻度角化过度,颗粒层减少,棘层变薄,真皮多无明显炎性细胞浸润。本例患者皮损为多发色素沉着性损害,无明确家族史,且其他系统疾病史,故应注意长期临床随访,排除肿瘤及其他系统疾病的发生。
本病临床上需要与花斑糠疹、玫瑰糠疹、固定性药疹、获得性鱼鳞病、单纯糠疹、大斑块型副银屑病、红斑期蕈样肉芽肿等相鉴别,主要根据各自相应特征性临床表现,结合组织病理学以及真菌学检查等。其中因蕈样肉芽肿属低度恶性皮肤T细胞淋巴瘤,需高度警惕。蕈样肉芽肿红斑期组织病理改变主要表现为真皮乳头及浅层T淋巴细胞为主的单一核细胞浸润,并可见淋巴细胞亲表皮现象[7]。本病目前尚无明确有效治疗方案,常选择外用糖皮质激素制剂、尿素、焦油、润肤剂等,外用或口服维A酸类药物也有一定效果,也有高浓度他卡西醇软膏外用成功治疗的报道[8]。如患者有系统疾病,积极治疗系统疾病有助于皮损消退。
猜你喜欢
中风与神经疾病杂志(2022年3期)2022-11-25
中国防痨杂志(2022年2期)2022-11-24
肝博士(2022年3期)2022-06-30
中国当代医药(2022年8期)2022-04-12
中国听力语言康复科学杂志(2021年6期)2021-12-21
数学学习与研究(2021年18期)2021-08-06
皮肤病与性病(2021年3期)2021-07-30
学习与科普(2019年36期)2019-09-10
风湿病与关节炎(2018年1期)2018-02-11
东方教育(2017年14期)2017-09-25
