
水液相环境下α-丙氨酸Mn(Ⅱ)配合物旋光异构的理论研究
2021-12-02 01:09刘军姜春旭刘芳高峰张雪娇雷泽萍佟华王佐成
浙江大学学报(理学版) 2021年6期
刘军,姜春旭,刘芳,高峰,张雪娇,雷泽萍,佟华,王佐成
(1.白城师范学院 计算机科学学院,吉林 白城 137000;2.白城师范学院 理论计算中心,吉林 白城 137000)
0 引言
α-丙氨酸(α-Ala)是蛋白质的基本单元,是生物体内的重要配体。按旋光性分为左丙氨酸(LAla)和右丙氨酸(D-Ala),按构型分为S丙氨酸(SAla)和R丙氨酸(R-Ala)。α-Ala 的手性特征使其金属配合物也具有手性。生命体内L-Ala 有活性,而D-Ala 太多会引发疾病[1-3]。锰是生命体必需元素,生命体内的锰一般为二价锰(Mn2+),其代谢失调可引起老年痴呆[4]。Mn2+可与某些肽键的羰基和亚氨基配位改变蛋白质结构[5]。氨基酸Mn2+螯合物对作物生长等有积极作用[6]。因此,对手性分子消旋反应的研究极为重要。
人们对α-Ala 及其金属配合物旋光异构进行了大量研究。文献[7-12]的研究表明,气相α-Ala 不能消旋,在水分子(簇)催化下α-Ala 可微量消旋,在水液相下α-Ala 可少量消旋,羟基负离子可促进α-Ala 的消旋反应。文献[13]的研究表明,SWBNNT(5,5)对α-Ala 的消旋具有明显的限域催化作用;文献[14]的研究表明,在MOR 分子筛限域下,羟自由基水分子簇的催化可使α-Ala缓慢消旋。另外,孤立的α-Ala与Cu2+、Ca2+、Na+、Zn2+、Mn2+、Mg2+、K+及Fe2+配合物不能消旋[15-22],在水液相下α-Ala·Zn2+和α-Ala·Fe2+能痕量消旋,可安全地用于生命体[23-24]。
为满足生命体的需求,细胞从细胞外摄取金属离子,但自由的金属离子对细胞有毒性,且细胞总是选择吸收金属离子的生物分子螯合物[25]。因不同旋光性的手性分子有不同的生物效应,α-Ala·Mn2+的优构体在生命体内的消旋情况关乎健康。考虑生命体的水环境分布较广,而水液相下α-Ala·Mn2+的消旋研究鲜见报道,基于已有研究[15-24],本文研究了水液相下α-Ala·Mn2+的消旋机理,探究了α-Ala·Mn2+用于生命体补充锰及丙氨酸的安全性。
1 研究与计算方法
采用有较高精度的泛函M06 方法[26],及自洽场理论的溶质全电子密度溶剂化(solvation model based on desity,SMD)模型[27],在6-31++G(d,p)基组下优化α-Ala·Mn2+旋光异构反应6 重态(在自旋态2,4,6 和8 中,6 重态最稳定)势能面上的驻点结构;采用自然键轨道(natural bond orbital,NBO)方法计算体系的自然布居分析(natural population analysis,NPA)电荷;通过对过渡态[28]做IRC(内禀反应坐标计算)分析[29],确认过渡态可信度。为得到较可靠的反应过程势能面,采用更高精度的明尼苏达泛 函 2015(minnesota nonseparable 2015,MN15)方 法[30],在高水平基组6-311++G(3df,2pd)下计算单点能。总吉布斯自由能是自由能热校正(自由能矫正在298.15 K 下进行)和单点能之和。S-Ala·Mn2+记作S-A·Mn,其在a 通道异构的第1 个S-型过渡态记作S-T1a,第1 个S-型中间体记作S-I1a,a 和b 通道共用结 构记作Xa(b);a 通道 上,4 个H2O 分子与Mn 配位、二聚水与S-A·Mn 氢键作用的体系记作S-A·Mn←4H2O·(H2O)2,其他体系表示方法相似。采用Gaussian 16[31]程序进行计算。
2 结果与讨论
将气相下A·Mn 的构象[19]作为初始猜测,优化得到A·Mn 的对映体S-A·Mn 和R-A·Mn,见图1。

图1 配合物S-A·Mn 及其手性对映体R-A·Mn 的几何构型Fig.1 Geometric conformation of complex S-A·Mn and chi‑ral enantiomer R-A·Mn
研究表明,S-A·Mn 可通过以O 为桥、顺次以O和N 为桥、以N 为桥及以Mn 为桥等的H 迁移实现旋光异构,分别命名为a,b,c 和d 通道。在水液相下有隐性和显性水溶剂效应,对于H 迁移反应,应考虑显性水溶剂效应,即水分子做H 迁移媒介、水分子与Mn 配位及水的极性溶剂作用;对于非H 迁移反应,只需考虑隐性水溶剂效应。为得到水液相下简洁、全面、清晰的S-A·Mn 旋光异构机理,分别对隐性和显性水溶剂效应进行讨论。
2.1 隐性水溶剂效应下S-A·Mn 的旋光异构
a 和b 通道共用前2 个基元,α-H 从α-C 羰基O迁移,c 和d 通道共用前3 个基元,S-α-Ala 从两性向中性异构。
2.1.1S-A·Mn 在a 和b 通道的手性对映体转变
S-A·Mn 在a 和b 通道的旋光异构历程见图2,反应的吉布斯自由能势能面见图3。

图2 S-A·Mn 在a 和b 通道的旋光异构历程Fig.2 Reaction process of optical isomerization of S-A·Mn in channel a and b

图3 S-A·Mn 在a 和b 通道旋光异构反应的吉布斯自由能势能面Fig.3 Gibbs free energy surfaces of optical isomerization of S-A·Mn in channel a and b
第1基元,S-A·Mn经14Mn和S-A上下相对运动的过渡态S-T1a(b),14Mn与S-A 的2个O 配位,异构成S-I1a(b)。从S-A·Mn 到S-T1a(b),14Mn—8O 键 长从0.207 3 nm 小幅拉伸至0.216 2 nm,14Mn—9O 键长从0.321 3 nm 收缩至0.252 0 nm,形成弱配位键,释放能垒,因此S-T1a(b)产生的能垒很低,仅为2.3 kJ·mol−1。
第2 基元,S-I1a(b)经2H 从1C 向8O 迁移的过渡态T2a(b)异构成I2a(b)。从S-I1a(b)到T2a(b),1C—2H键长从0.109 6 nm 拉伸至0.151 9 nm,7C—8O 双键长从0.126 6 nm 拉伸至0.131 2 nm,14Mn—8O 键长从0.221 1 nm 拉伸至0.322 6 nm,这些变化所需能量很多,T2a(b)产生254.8 kJ·mol−1的能垒,该能垒小于气相下S-A·Mn 旋光异构此基元的能垒293.9 kJ·mol−1[19],水溶剂对此基元有催化作用。原因是水液相下S-I1a(b)的偶极矩(7.295 3D)小于T2a(b)的偶极矩(14.336 0D),极性水溶剂的作用使T2a(b)变得相对稳定。
I2a(b)的异构 分为a 和b 两个 分通道。
(1)a 分通道第3 基元,I2a(b)经与T2a(b)镜像对称的过渡态T3a,2H 在纸面内侧从8O 迁移至1C,异构成R-I3a,S-A·Mn 实现旋光异构。从I2a(b)到T3a,8O—2H 键长从0.096 9 nm 拉伸至0.119 2 nm,断裂,T3a产生158.8 kJ·mol−1的能垒。
第4 基元,R-I3a经 与S-T1a(b)镜像对称的过渡态R-T4a,14Mn 相 对于S-A 向下 运动,异构成RA·Mna。从R-I3a到R-T4a,14Mn—9O 键长从0.237 2 nm 小幅拉伸至0.252 0 nm,所需能量很少,14Mn—8O 键长从0.221 1 nm收缩至0.216 2 nm,释放能垒,因此R-T4a产生的能垒极低,只有1.7 kJ·mol−1。结构分析表明,R-A·Mna与S-A·Mn 呈镜像对称,S-A·Mn 实现了手性对映体转变。
(2)b 分通道第3 基元,I2a(b)经5H 在纸面内侧从3N 向1C 迁移的过渡态T3b异构成R-I3b,S-A·Mn 实现旋光异构。从I2a(b)到T3b,3N—5H 键长从0.102 7 nm 拉伸至0.123 9 nm,断裂,T3b产生167.8 kJ·mol−1的能垒。
第4 基元,R-I3b经4H 和6H 左右翻转的过渡态R-T4b异构成R-I4b。氨基翻转所需能量不高,RT4b产生11.6 kJ·mol−1的能垒。
R-I4b的异构分为b1 和b2 两个路径。
(1)b1 路径第5 基元,R-I4b经14Mn 相 对7C—9O 键左右翻转的过渡态R-T5b1,14Mn 从羧基内侧翻转到外侧,与3N、1C、7C 和9O 形成螯合环,异构成R-I5b1。从R-I4b到R-T5b1,丙氨酸与Mn 相对运动,所需能量很少,R-T5b1产生的能垒仅为8.5 kJ·mol-1。
第6 基元,R-I5b1经8O—7C 键仰视顺(或逆)时针内旋转的过渡态R-T6mb1(或R-T6nb1)异构成较稳定的中性丙氨酸与二价锰的配合物R-A*·Mnb1。从R-I5b1到R-T6mb1,8O—7C 键旋转94.0°,RT6mb1产 生32.3 kJ·mol−1的能垒;类似地,R-T6nb1产生32.7 kJ·mol−1的能垒。
(2)b2 路径第5 基元,R-I4b经7C—1C 键右视顺时针内旋转的过渡态R-T5b2异构成R-I5b2。从R-I4b到R-T5b2,10C—1C 键内旋转92.4°,R-T5b2产生的能垒为7.9 kJ·mol−1。
第6 基元,R-I5b2经2H 从8O 向3N 迁移的过渡态R-T6b2异构成两性丙氨酸与锰的配合物R-A·Mnb2。从R-I5b2到R-T6b2,8O—2H 键长从0.100 0 nm 拉伸至0.111 8 nm,断裂;R-I5b2的8O—2H 键被活化(键长为0.100 0 nm);2H 逆着体系偶极矩的方向运动,同时体系电场力助力3N 迁移,R-T6b2产生的能垒极小,仅为1.3 kJ·mol−1。分析表明,S-A·Mn 实现了手性对映体转变。
由图3 可知,S-A·Mn 在a 和b 通道消旋决速步共用第2 基元,内禀能垒为255.4 kJ·mol−1。由于当正负反应能垒在40.0 kJ·mol−1以下时反应物和产物可以共存[32],因此,反应物S-A·Mn 和S-I1a(b)共存,前者的分布高;产物R-A·Mna(b2)、R-I3a、R-I5b2、RA*·Mnb1以及R-I5b1共存,R-A·Mna(b2)的分布最高,R-I3a次之,R-A*·Mnb1第3。
2.1.2S-A·Mn 在c 和d 通道的手性转变
S-A·Mn 在c 和d 通道的旋光异构历程见图4,反应的吉布斯自由能势能面见图5。

图4 S-A·Mn 在c 和d 通道的旋光异构历程Fig.4 Reaction process of optical isomerization of S-A·Mn in channel c and d
第1 基元,S-A·Mn 经H 在3N 和9O 间 迁移的过渡态S-T1c(d),6H 从3N 迁移至9O,异构成SI1c(d)。从S-A 到S-T1c(d),3N—6H 键长从0.102 4 nm 拉伸至0.144 0 nm,反应物的3N—6H 键已经活化,其断裂所需能量不多,S-T1c(d)产生的 能垒为32.4 kJ·mol−1。
第2 基元,S-I1c(d)经9O—7C 键内旋转的过渡态S-T2mc(d)或S-T2nc(d),6H 绕9O—7C 键俯视逆时针(或顺时针)旋转,6H 从羧基外侧转到内侧,异构成S-I2c(d)。从S-I1c(d)到S-T2mc(d),9O—7C 键内旋转90.6°,所需能量不多,但此过程6H 的运动方向与体系偶极矩的方向始终成锐角,需额外给予能量抵抗体系电场力做功,所以S-T2mc(d)产生的内禀能垒不会太低,为57.1 kJ·mol−1;类似地,S-T2nc(d)产生的能垒为61.1 kJ·mol−1。
第3 基元,S-I2c(d)经4H 和5H 在纸面内外翻转的过渡态S-T3c(d),4H 和5H 从纸面外进入纸面内,异构成S-I3c(d)。从S-I2c(d)到S-T3c(d)是非骨架异构,所需能量很少,S-T3c(d)产生的内禀能垒是11.4 kJ·mol−1。此时氨基氮3N 正面裸露,电子云密度增大,为α-H 向其迁移创造了空间条件。
S-T3c(d)的 异构分为c和d 两个分通道。
(1)c 分通道 第4 基 元,S-I3c(d)经2H 从1C 向3N迁移的过渡态T4c异构成I4c。从S-I3c(d)到T4c,1C—2H 键长从0.110 0 nm 拉伸至0.128 4 nm,断裂,键角3N—1C—10C—7C 从128.4°增至155.6°,T4c产生的能垒为220.8 kJ·mol−1,此能垒远小于气相下此基元反应的能垒331.8 kJ·mol−1[19]。原因在于气相下此基元反应是H 从α-C 向氨基N 迁移与H从N 向O 迁移协同进行的双质子迁移过程。
抗震设防烈度为8度,按罕遇地震设计基本地震加速度值为0.20g,设计地震属于第一组,场地类别为Ⅱ类,则Tg=0.4s。阻尼比取0.02,地震影响系数曲线的阻尼调整系数按1.0采用,竖直地震载荷对整个结构的动力响应影响较小,一般情况下,只考虑水平向地震作用[5],故分别在结构的两个水平主轴方向计算地震作用。由图3求得加速度谱值如表2。
第5 基元,I4c经 与T4c镜像对称的过渡态T5c,实现了5H 从3N 在纸面内侧向1C 的迁移,异构成R-I5c,S-A·Mn 实现旋 光异构。从I4c到T5c,3N—5H 键长从0.102 7 nm 拉伸至0.124 0 nm,T5c产生的能垒为168.9 kJ·mol−1。
第6 基元,R-I5c经 与S-T3c(d)镜像对称的过渡态R-T6c,实现了2H 和4H 从右侧偏前的方位向纸面内侧翻转,异构成R-I6c,为6H 从羧基内侧向外侧旋转创造了空间条件。R-T6c产生的能垒为13.8 kJ·mol−1。
第7 基元,R-I6c经 与S-T2nc(d)和S-T2mc(d)镜像对称的过渡态R-T7mc和R-T7nc,6H 从羧基内侧向外侧旋转,异构成R-I7c。从R-I6c到R-T7mc或R-T7nc,只是化学键内旋转,所需能量较少,RT7mc和R-T7nc产生的能垒分别为35.8 和339.8 kJ·mol−1,低于第2 基元的能垒。原因在于此基元反应6H 是逆着偶极矩方向运动的,体系电场力助力6H 从羧基内侧向外侧旋转。分析表明,R-I7c同于R-I5b2,其在第8 基元的异构同于R-I5b2的异构,得到产物R-A·Mnb2(c)。
(2)d 分通道第4 基元,S-I3c(d)经14Mn 相 对8O—7C 键左右翻转的过渡态S-T4d,14Mn 从羧基内侧翻转至外侧,异构成S-I4d。从S-I3c(d)到S-T4d,键角14Mn—8O—7C 从126.6°增至171.2°,S-T4d产生的能垒为4.6 kJ·mol−1。
第5 基元,S-I4d经2H 在1C—14Mn 键迁移的过渡态T5d,2H 从1C 迁移至14Mn,异构成I5d。从SI4d到T5d,1C—2H 键长从0.109 9 nm 大幅拉伸至0.267 6 nm,键角3N—1C—10C—7C 从127.5°增至169.8°,这些变化所需能量很多,T5d产生的能垒为293.3 kJ·mol−1,该能垒远大于气相下此基元的能垒5.3 kJ·mol−1[19]。原因有二,一是气相下对应的此基元反应物的C—H 键长为0.123 2 nm,被较好地活化,红外振动频率为1 348.6 cm−1,而本文中水液相下此基元1C—2H 键长为0.109 9 nm,红外振动频率为3 048.7 cm−1,说明水溶剂效应使1C—2H 键严重钝化;二是水液相下的偶极矩(17.047 1D)远大于T5d的偶极矩(12.309 9D),极性水溶剂的作用使T5d较S-I4d更不稳定。
第6 基元,I5d经2H 在纸面内外翻转的过渡态T6d,2H—14Mn—8O 从纸面外侧翻转到内侧,异构成I6d。从I5d到T6d,7C—1C 键内旋转13.5°,键角14Mn—8O—7C—9C 从168.6°增 至180.0°,键角2H-14Mn—8O—7C 从−45.8°增至0°,这些微小的变化所需能量很少,T6d产生的能垒为2.9 kJ·mol−1。
第7 基元,I6d经过渡态T7d(与T5d镜像对称),2H 在纸面内侧从14Mn 迁移至α-碳1C,异构成RI7d,S-A·Mn 实现旋光异构。从I6d到T7d,Mn—H键长基本没变,骨架二面角14Mn—8O—7C—1C 从11.3°增至37.2°,键角2H—14Mn—8O—7O 从45.8°减至4.8°,键角2H—14Mn—8O 从110.6°减至87.0°,这些变化使T7d产生72.3 kJ·mol−1的能垒。
第8基元,R-I7d经过渡态R-T8d(与S-T4d镜像对称),14Mn从羧基外侧翻转至羧基内侧,异构成R-I8d。从R-I7d到R-T8d,键角14Mn—8O—7C 从125.2°增至171.2°,R-T8d产生的能垒为10.6 kJ·mol−1。
分析表明,R-I8d同于R-I5c,即R-I8d的异构同于R-I5c向R-A·Mnc的异构,c 通道的第6,7 和8 基元反应即为d 通道的第9,10 和11 基元反应,不再赘述。
由图5 可知,c 通道旋光异构决速步是第4 基元,能垒为220.8 kJ·mol−1,d 通道旋光异构决速步是第5 基元,能垒为293.3 kJ·mol−1。综合图2~图5可知,当只考虑隐性水溶剂效应时,S-A·Mn 在c 通道的旋光异构最具优势,决速步能垒来自α-H 从α-C 向N 迁移的过渡态;a 和b 通道为亚优势通道,决速步能垒来自α-H 从α-C 羰基O 迁移的过渡态;d通道为劣势通道,决速步能垒来自α-H 从α-C 向Mn 迁移的过渡态。220.8 kJ·mol−1的能垒远高于反应极限能垒167.0 kJ·mol−1[32],隐性水溶剂效应下丙氨酸二价锰配合物不能旋光异构。与S-A·Mn 在气相下旋光异构反应通道[19]的优劣顺序相比,隐性水溶剂效应使反应通道的优劣顺序发生改变,气相下的优势通道在液相下变为劣势通道。
2.2 显性水溶剂效应下S-A·Mn 的手性转变
已有研究[10-12]表明,对于α-C 与N 间以及α-C与O 间的H 转移,二聚水和三聚水的催化作用差别较小,单个水分子的催化作用远小于二聚水和三聚水,且液相下的水分子绝大多数以水簇的形式存在。为使问题简便且节省篇幅,仅讨论二聚水作H迁移媒介的决速步。配位键强于氢键,水分子(簇)优先与Mn2+配位,再与体系形成氢键。
2.2.1 水分子(簇)对a 和b 通道决速步的影响
a 和b 通道旋光异构的决速步都是第2 基元,水分子(簇)与中间体反应物S-I1a(b)的作用有2 种,一是H2O 与Mn2+形成配位键,二是(H2O)2与S-I1a(b)的2H 和8O 形成氢键。Mn2+与O 最多可六配位,H2O 与Mn2+配 位饱和后 与S-I1a(b)氢键 作用。Mn2+与O 已是二配位,再与4 个H2O 配位,形成S-I1a(b)←4H2O。(H2O)2再 与S-I1a(b)←4H2O 的2H、8O 和3N氢键作用,同时与S-I1a(b)←4H2O 的25H—24O—26H 形成氢键网络,记作S-I1a(b)←4H2O·(H2O)2。(H2O)2从1C 向8O 迁移传递α-H 的历程见图6(a),反应的势能面见图7(a)。
S-I1a(b)←4H2O·(H2O)2经H 迁 移、骨架形变、水分子间及水分子与氨基间氢键网络破裂协同进行的过渡态T2a(b)←4H2O·(H2O)2,实现H 从1C 向8O的净迁移,异构成I2a(b)←4H2O·(H2O)2。从S-I1a(b)←4H2O·(H2O)2到T2a(b)←4H2O·(H2O)2,1C—2H键长从0.109 6 nm 拉伸至0.181 8 nm,断裂,27O—29H 键长从0.098 1 nm 拉伸至0.128 5 nm,断裂,30O—32H 键长从0.098 0 nm 拉伸至0.102 8 nm,断裂,键角3N—1C—10C—7C 从120.3°增至141.3°,稳定的氢键网络被严重破坏,这些变化使T2a(b)←4H2O·(H2O)2产生165.8 kJ·mol−1的能垒,该能垒远小于T2a(b)产生的能垒254.8 kJ·mol−1,水分子(簇)起了很好的催化作用。原因在于八元环过渡态T2a(b)←4H2O·(H2O)2的3 条氢键的键角1C—2H—27O、27O—29H—30O 和30O—32H—12O 接近平角,氢键很强,T2a(b)←4H2O·(H2O)2的稳定性较好。165.8 kJ·mol−1高于水液相下二聚水作H 迁移媒介丙氨酸旋光异构在此基元的能垒141.8·mol−1[12],说明Mn2+对此基元反应起了阻碍作用。原因在于水液相下Mn2+与H2O 配位形成的S-I1a(b)←4H2O·(H2O)2有稳定的氢键网络,从S-I1a(b)←4H2O·(H2O)2到T1a(b)←4H2O·(H2O)2,除提供碳氢键、氧氢键断裂和骨架形变所需的能量外,还需提供破坏氢键网络的能量。
2.2.2 水分子(簇)对c 通道决速步的影响
c 通道旋光异构的决速步是第4 基元,S-I3c的Mn 与5 个H2O 配位(Mn 已与一个O 配位),形成SI3c←5H2O。(H2O)2再与3N、2H 以及S-I3c←5H2O中的H2O 氢键作用,形成具有稳定氢键网络的中间体S-I3c←5H2O·(H2O)2。(H2O)2从1C 向3N 迁移传递H 的历程见图6(b),反应的势能面见图7(b)。
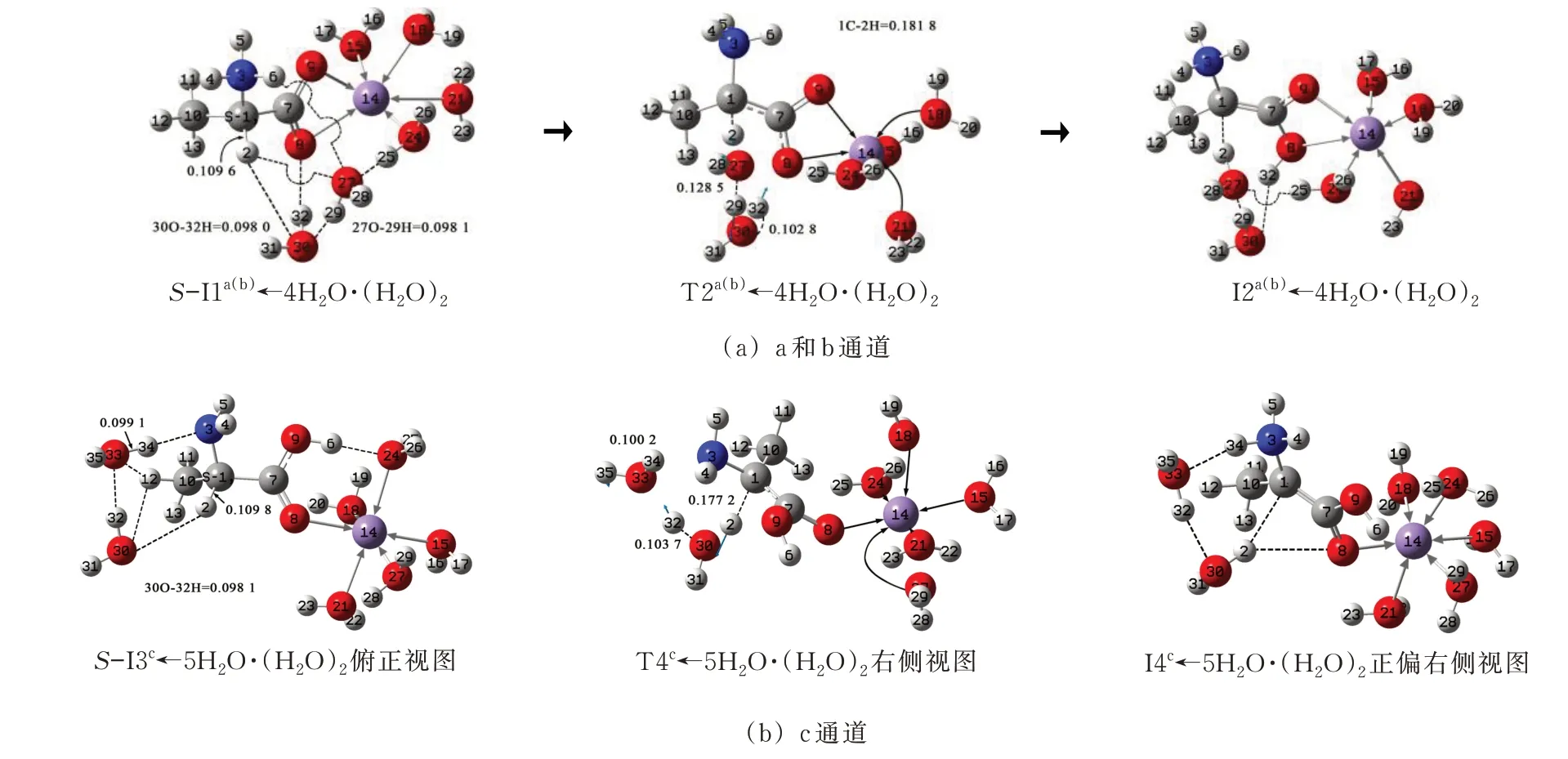
图6 水分子(簇)作用下S-A·Mn 在a,b 和c 通道旋光异构反应的决速步(虚线表示氢键作用)Fig.6 The rate-determining step of optical isomerization of S-A·Mn in channel a,b and c under the action of water molecule(cluster)

图7 水分子(簇)作用下S-A·Mn 在a,b 和c 通道旋光异构反应决速步的势能面Fig.7 Potential energy surfaces of optical isomerization of S-A·Mn in channel a,b and c under the action of water molecule(cluster)
S-I3c←5H2O·(H2O)2经3H 迁移及氢键网络被破坏协同进行的过渡态T4c←5H2O·(H2O)2,异构成I4c←5H2O·(H2O)2。从S-I3c←5H2O·(H2O)2到T4c←5H2O·(H2O)2,1C—2H 键 长从0.109 8 nm 拉伸至0.177 2 nm,30O—32H 键长从0.098 1 nm 拉伸至0.103 7 nm,33O—34H 键长从0.099 1 nm 拉伸至0.100 2 nm,键角3N—1C—10C—7C 从125.9°增至167.9°,24O—6H、30O—12H 及33O—12H 间的氢键断裂,这些变化使T4c←5H2O·(H2O)2产生155.1 kJ·mol−1的能垒,与隐性水溶剂效应下T4c产生的内禀能垒(220.8 kJ·mol−1)相比有大幅下降,(H2O)2对此基元反应起了很好的催化作用。T4c←5H2O·(H2O)2产生的内禀能垒与显性水溶剂效应下丙氨酸旋光异构过程中此基元反应的决速步能垒(约为110.0 kJ·mol−1[11])相比有显著增加,说明Mn2+对此基元反应有负催化作用。原因在于Mn2+的存在使此基元的中间体反应物形成了稳定的氢键网络,从S-I3c←5H2O·(H2O)2到T4c←5H2O·(H2O)2,破坏氢键网络需额外给予能量。
2.2.3 水分子(簇)对d 通道决速步的影响
d 通道旋光异构的决速步是第5 基元,S-I4d的Mn 与5 个H2O 配位达到满配状态(Mn 已与一个O配位),形成S-I4d←5H2O。水分子(簇)(为简单起见,只考虑1 个H2O)再与S-I4d的2H 氢键作用,同时与Mn 配位的水分子氢键作用,形成稳定的氢键网络(图8)。由图8 可知,14Mn 已经无“能力”再接收质子,亦即α-H 无法通过水分子作媒介从α-C 向Mn 迁移。因此,在真正意义的水液相环境下(考虑水分子的作用),S-A·Mn 无法在d 通道实现旋光异构。
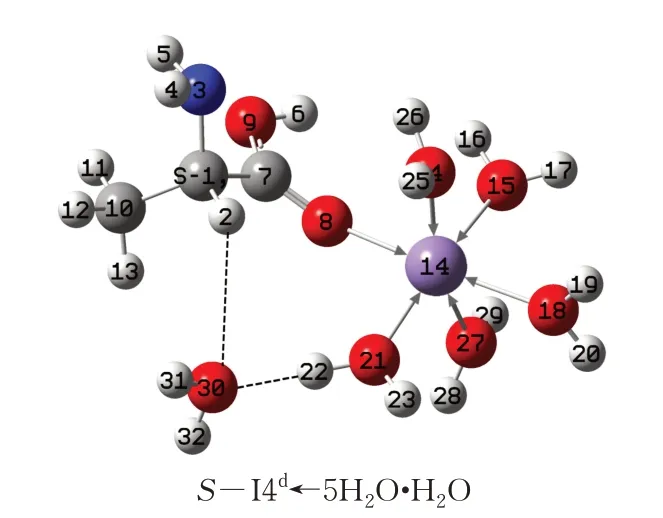
图8 水分子与S-I4d作用形成的具有稳定氢键网络体系的复合物Fig.8 Compound with stable hydrogen bond network formed by interaction of water molecule and S-I4d
综上可知,在显性水溶剂效应下,S-A·Mn 在c通道的旋光异构最具优势,决速步自由能垒为155.1 kJ·mol−1;亚优势通道是a 和b,决速步自由能垒 为165.8 kJ·mol−1;d 是 劣势通道,S-A·Mn 无法在该通道旋光异构。S-A·Mn 在c 通道上旋光异构的决速步能垒为155.1 kJ·mol−1,接近反应极限能垒167.0 kJ·mol−1[32],因此在水液相下S-A·Mn 只能微量消旋,A·Mn 用于生命体补充Mn2+和丙氨酸具有很好的安全性。
3 结论
在MN15/SMD/6-311++G(2df,pd)//M06/SMD/6-31++G(d,p)双水平下,考查了在水液相下S-A·Mn 在a,b,c,d 4 个通道的旋光异构,a 通道H 以O 为桥迁移,b 通道H 分别顺次以O 和N 为桥迁移,c 通 道H 以N 为桥迁 移,d 通道H 以Mn 为桥迁移,得到如下结论:
3.1 在隐性水溶剂效应下,优势通道c 的决速步能垒为220.8 kJ·mol−1,来自从α-C 向N 迁移的α-H 过渡态;亚优势通道a 和b 的决速步能垒为254.8 kJ·mol−1,来自 从α-C 向O 迁移的α-H 过渡态;劣势通道d 的决速步能垒为293.3 kJ·mol−1,来自从α-C 向Mn 迁移的α-H 过渡态。
3.2 在显性水溶剂效应下,c 通道决速步能垒降至155.1 kJ·mol−1,a 和b 通道决速步能垒降至165.8 kJ·mol−1,d 通道无法实现旋光异构。
3.3 在水液相环境下,S-A·Mn 只能微量消旋,A·Mn 用于生命体补充Mn2+和丙氨酸具有很好的安全性。
猜你喜欢
河北师范大学学报(自然科学版)(2022年5期)2022-09-20
兵工学报(2022年2期)2022-05-22
原子与分子物理学报(2022年3期)2022-03-05
波谱学杂志(2021年3期)2021-09-07
辽宁科技大学学报(2021年2期)2021-07-22
兵工学报(2021年4期)2021-06-19
——以高中化学“氢键”的教学为例
教学月刊(中学版)(2020年13期)2020-12-29
兵工学报(2020年12期)2020-02-06
青岛大学学报(工程技术版)(2019年2期)2019-09-10
科学导报(2018年30期)2018-05-14
