
Phonon dispersion relations of crystalline solids based on LAMMPS package∗
2021-11-23 07:28ZhiyongWei魏志勇TianhangQi戚天航WeiyuChen陈伟宇andYunfeiChen陈云飞
Chinese Physics B 2021年11期
关键词:陈云
Zhiyong Wei(魏志勇) Tianhang Qi(戚天航) Weiyu Chen(陈伟宇) and Yunfei Chen(陈云飞)
1Jiangsu Key Laboratory for Design&Manufacture of Micro/Nano Biomedical Instruments and School of Mechanical Engineering,Southeast University,Nanjing 211189,China
2College of Mechanical and Electronic Engineering,Nanjing Forestry University,Nanjing 210037,China
Keywords: phonon dispersion relation,molecular dynamics,force constant,potential function
1. Introduction
Phonons are collective excitations and main energy carriers in crystalline solids, and also have important effects on many physical properties.[1,2]For example,phonon dispersion and scattering can determine the thermal conductivity of insulators and semiconductors.[3-5]When two solids come together to form an interface, the overlap of the phonon density of states between the two solids can influence the heat flux across the interface.[6-8]In addition, many other physical properties of solid, such as the sound velocity and the elastic constants, can be obtained from the low-frequency region of the phonon dispersion relation.[9]Several recently found physical phenomena or results are also closely related to phonon dispersion, such as hydrodynamic phonon transport,[10]curvature-related phonon transport,[11]and resonant scattering.[12]Harmonic lattice dynamics is a simple model that approximates the interactions among atoms as springs without considering the higher-order interactions,which have been established in the 1920s and 1930s by Bonn and Huang.[13]From the harmonic lattice dynamics theory,one can calculate the frequency and eigenvector of any phonon mode in the first Brillouin zone(FBZ)of the crystal,and can further obtain the phonon group velocity,capacity,irradiation heat flux,[14]etc. The optical phonon modes at some particular location (such as the center or boundary) of the FBZ would be relevant to the IR or Raman information.[15]Therefore, the rapid and accurate acquisition of phonon dispersion relations of materials or structures is of great significance for understanding the mechanical,thermal,and optical properties of solids.
The General Utility Lattice Program(GULP)package[16]is a commonly used tool to calculate the phonon properties of crystalline solids. This package can use a large number of potential functions or force constants as input to obtain the phonon dispersion relation. However, all the internal functions of this software package are integrated together, which makes it difficult for researchers to modify the code to adapt to some new potential functions. The Phonopy package[17]is another commonly used tool to calculate the phonon properties of crystalline solids. This package can use force constants obtained from various external packages, including first principle packages (VASP[18]and QUANTUM ESPRESSO,[19]etc.) and MD package(LAMMPS),as input to get the phonon dispersion relation. However,the interfaces between Phonopy and these external packages are not easy to set up, which needs to prepare many intermediate process files. Since the LAMMPS package[20]is widely used to simulate phonon or thermal transport of crystalline solids, the MD simulation results,such as the thermal conductivity and the thermal boundary conductance, can be self-consistently interpreted if the phonon dispersion relations of the structure are obtained directly using the same potential function in the LAMMPSbased MD simulation. As far as we know, a command to calculate phonon dispersion relation has been included in the LAMMPS package,[21]but it requires constructing a large simulation system to accommodate the number of wave vectors in the FBZ.If the simulation time is short,the calculation error is also large. In addition,when the size of the unit cell is large,such as phononic crystals,the calculation is often timeconsuming.
In this study, a new tool based on the LAMMPS package is proposed to obtain the phonon dispersion relation of crystalline solids according to the harmonic lattice dynamics principle. The main advantage of this approach is that any potential function or force field that is integrated with the LAMMPS package can be used to calculate the phonon dispersion relation corresponding to it, and the calculation is much faster even for the large unit cells. In the second section, we present the detailed method and calculation process to obtain the phonon dispersion relation based on the LAMMPS package,including the acquisition of force constant,the construction of dynamic matrix, and so on. The phonon dispersion relations of several typical materials are demonstrated in the third section. The relevant home-made code and script can be found in the supporting information.
2. Method
Suppose a simulation system containsLunit cells, and each unit cell containsbatoms. Then, there are totallyL×batoms in the simulation system.When all the atoms in the system are in an equilibrium state, the applied force on thej-th atom,Fj,is

Here,Fjiis the applied force ofi-th atom on thej-th atom,kjipresents the interaction force constant matrix betweenj-th andi-th atoms,rjiis the relative displacement betweenj-th andi-th atoms. When the simulation system reaches the minimum potential energy point,the force applied to each atom is zero. Under this condition, if moving thei-th atom by a tiny distance dxalong thexdirection while keeping the positions of all other atoms still in their equilibrium positions,then the changed force ∆Fjon thej-th atom can be written as

Thus,the spring stiffness componentskjixx,kjiyx,kjizxbetweenj-th andi-th atoms can be obtained as
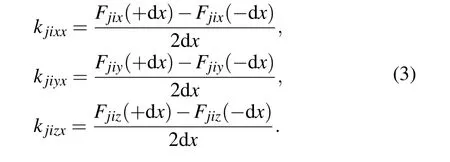
In the same way,the remained six spring stiffness components can also be obtained by moving thei-th atoms along theyandzdirections,respectively. Due to the translation symmetry of the crystal,the force constant between thei-th atom and itself can be obtained by the following equation:[22]

In the LAMMPS package, we can easily move the displacement of atoms one by one in a specified unit cell and calculate the force on all the other atoms using a loop script. The corresponding LAMMPS script can be found in the supporting information. Thus, we can quickly obtain the force constant matrix between any pair of atoms corresponding to the used potential functions with the help of the LAMMPS package.
After obtaining all the force constant matrices, the dynamics matrixDof the simulation system can be constructed from the lattice dynamics theory as


Figure 1 shows the flow chart for calculating the phonon dispersion relation based on the LAMMPS package. First,the simulation system should be prepared with the same atomic order in each unit cell according to the equilibrium lattice parameters of the crystal. The equilibrium lattice parameters are equivalent to the equilibrium distances between neighboring atoms at the lowest potential energy in a crystal. Note that the equilibrium lattice parameter of one crystalline solid may be not the same when simulating with different potentials or force fields. For example,the predicted carbon-carbon(C-C)bond length from the Tersoff potential in 1988 is about 1.46 ˚A,while the corresponding length is 1.44 ˚A from the Tersoff potential in 2010.[23]Both of them are greater than the experimentally observed value of 1.42 ˚A. In order to obtain the equilibrium lattice parameters for the specified potential,one can calculate the potential energy of the simulation system as a function of the lattice parameter. The equilibrium lattice parameter is the value corresponding to the minimum potential energy. Then,a home-made LAMMPS script(see the supporting information(SI))is run to calculate the force on each atom when moving the position of the atoms of the first unit cell one by one with tiny displacement. The typical tiny displacement ranges from 0.01 to 0.001 ˚A.After running this script with the LAMMPS package,several files,including the force on each atom,can be obtained. With these files as the input,Eq.(3)is used to calculate the force constant matrix between any pair of atoms,and Eq. (4) for the force constant matrix between one atom and itself according to the translational invariance of the crystal.The relevant code that deals with the output files of LAMMPS and calculates the force constant matrix can be found in the SI.After all the force constants are determined,Eq.(5)is used to construct the dynamics matrix. Lastly,after a series of phonon wavevectors are given, the eigenvalues (or the square of the phonon frequency) of the dynamic matrix corresponding to the wavevector can be solved numerically. The relation between phonon frequency and phonon wavevector, namely the phonon dispersion relation,can be obtained.The relevant code that constructs the dynamics matrix and obtains the phonon dispersion can be found in the SI.

Fig.1. The flow chart for the calculation of phonon dispersion of crystals. The given input script for LAMMPS and the relevant codes(in the supporting information)are for the single-layer graphene model of two-atom unit cell. Some key parameters or details that need to change for other materials and structure have been explained in the comment lines of the script and codes.
Note that the basic theory to construct the dynamical matrix in Ref. [21] is different from this work, although both are from the LAMMPS package. The former is based on the fluctuation-dissipation theory. It requires a long equilibrium molecular dynamics simulation to obtain the atomic trajectories,and the effective phonon modes depend on the simulation size. Our method is based on the lattice dynamics theory. It only needs the force constants among atom pairs. Compared with Ref.[21],our method cannot identify the temperature effect on the dispersion,but the calculation time will be several orders of magnitude shorter because it does not need the long equilibrium molecular dynamics simulations.
3. Case studies
3.1. Phonon dispersion of graphene
Graphene is a two-dimensional crystalline solid that contains two atoms in the unit cell. The Tersoff potential in 2010,[23]which is fairly efficient to simulate the graphene in MD simulation due to its analytical forms,is first used to check the validity of the proposed technique for the calculation of phonon dispersion relation. First, the potential energy of every atom in the simulation system as a function of the lattice parameter is obtained in Fig. 2(a). It is found that the potential energy reaches the local minimum when the lattice parameter is 1.44 ˚A, indicating that the equilibrium lattice parameter is 1.44 ˚A. Then, an atomic model with the equilibrium lattice constantag=1.44 ˚A is prepared for the graphene, in which 64 atoms are large enough as long as the simulation system satisfies the periodic boundary conditions. The black solid lines in Fig. 2(b) present the calculated phonon dispersion relation of graphene according to the flow chart of Fig.1.Since the parameters of the Tersoff potential in 2010 are already optimized by Lindsay and Broido,the obtained phonon dispersion agrees well with that calculated from DFT(the red solid lines in Fig.2(b)). The phonon dispersion relation calculated from the proposed technique is almost the same as previous publications.[23]The phonon density of states is also very easy to obtain by sampling the phonon wavevectors uniformly throughout the FBZ and then calculating the corresponding phonon frequencies,as shown in Fig.2(c).

Fig. 2. Equilibrium lattice parameter (a), phonon dispersion relation(b)and vibrational density of states(c)of graphene calculated from the proposed method with the Tersoff potential in 2010. The black solid lines in(b)are the phonon dispersion of graphene from the DFT.
Although the MD simulation is often used to model the graphene as well as its composite due to their superior physical properties and potential applications, one of the most important problems in MD simulation is the low reliability of the calculation. Since the only uncertainty in MD simulation is the atomic interactions, the accuracy of MD depends directly on the selection of the potential function used in the simulation. There have been more than five different potentials in the LAMMPS package that can be used to model graphene.Since the phonon dispersion relation is an important factor determining the mechanical and thermal properties of materials,we will use the proposed method to calculate the phonon dispersion relation of graphene and evaluate the advantages and disadvantages of these potential functions.
Besides the above referred Tersoff potential in 2010,[23]other four commonly used potentials for MD simulation of the graphene in LAMMPS,including Tersoff potential in 1988,[23]AIREBO,[24]REAXFF,[25]and polymer consistent force field(PCFF),[26]are selected to calculate and compare the phonon dispersion relation of graphene. Tersoff potential in 1988 is mainly used to model the short-range covalent bond interactions among carbon atoms. Figure 3(a) shows that the obtained optical phonon frequency can be as high as 70 THz,which is much higher than the result of the DFT calculation(see the black solid lines in Fig.2(b)).Compared with the Tersoff potential, the AIREBO potential not only can model the short-range C-C covalent interactions but also can model the long-range interactions,such as the interlayer interactions between different graphene layers in multi-layer graphene film or bulk graphite. The obtained phonon dispersion relation from AIREBO,as shown in Fig.3(b),is also very close to the results of the DFT.However,one disadvantage of AIREBO potential is the longer simulation time. Our previous simulations have shown that the cost simulation time with the AIREBO potential is about six times longer than that with the Tersoff potential for the same three-dimensional graphene model.[27]The force field of REAXFF potential is fitted from the quantum mechanics calculation. The existed potential files in the LAMMPS can be used to model the atomic system that includes the C,O, Si, Al, etc. Figure 3(c) shows that the obtained phonon dispersion relation of graphene from REAXFF potential deviated greatly from the results of DFT,especially for the optical phonon and longitudinal acoustic phonon branches. In addition to the above three potentials, the PCFF potential is often used to model the graphene composite system[28]by dealing with atomic interactions as simple bond spring, angle spring,etc. Figure 3(d) shows that the optical phonon frequency atΓpoint from PCFF potential is slightly higher than the corresponding value from the DFT calculation. In addition, the predicted ZA phonon dispersion from PCFF potential is linear, which is also not consistent with the calculation of DFT.Therefore, it is suggested that the Tersoff potential in 2010(see Fig. 2(b)) is more suitable for simulating graphene according to the comparison between the phonon dispersion relation from the DFT calculation and those predicted by the five classical potential functions.

Fig. 3. Comparison of phonon dispersion of graphene from Tersoff (a) in 1988, AIREBO (b), REAXFF (c) and PCFF (d) potentials. These results are compared with that of DFT to easily evaluate the accuracy of the empirical potential functions.
3.2. Phonon dispersion of superlattice
In this section, the validity of the proposed technique is tested for phonon dispersion relation of the crystalline solids with the large unit cells,such as the phononic crystal or superlattice. Here,the in-plane and the cross-plane phonon dispersions of SinGensuperlattice are calculated using the proposed technique. To simplify the calculation, the Stillinger-Weber(SW)potential[29]for silicon is used to model the atomic interactions between all atoms. The masses of Si and Ge atom are set to 28 g/mol and 73 g/mol,respectively. Figure 4 shows the atomic structure of the SinGenwithn=1. The corresponding periodic length along the cross-plane direction isL=2n·asi,whereasiis the conventional unit cell size of bulk silicon.

Fig.4. The atomic model of SinGen superlattice with n=1. The corresponding periodic length along the cross-direction(or c-axis)is L=2n·asi.

Fig.5. The in-plane(a)and cross-plane(b)phonon dispersions of SinGen superlattice for the Si1Ge1,and the in-plane(c)and cross-plane(d)phonon dispersions for the Si10Ge10. The atomic numbers in the unit cell of the Si1Ge1 and Si10Ge10 are 16 and 160,respectively.
For the superlattice withL= 2asi, there are totally 16 atoms in the unit cell, and thus 48 phonon branches for each wavevector.Figures 5(a)and 5(b)show the calculated in-plane and cross-plane phonon dispersions,respectively. The results indicate that there are many phonon bandgaps above 10.5 THz along the cross-plane direction,while there is only one phonon bandgap around 11 THz along the in-plane direction. A similar technique is also performed to calculate the phonon dispersion relation of superlattice structures withL=20asias shown in Figs.5(c)and 5(d). The atom numberNin the unit cell of Si10Ge10isN=160. The running timetfor the LAMMPS script is aboutt=305 s in a laptop with an Intel single-core CPU@2.7 THz.Figure 6 presents the time needed to calculate the phonon dispersion of SinGensuperlattice with variable unit cell sizes using the same laptop. It indicates that the needed time is less than one minute when the atomic number in the unit cell is less than 100.For larger unit cells,parallel computing with multiple cores in LAMMPS can be used conveniently to reduce the running time. Therefore,the proposed tool also can be used for the fast calculation of the phonon dispersion with large unit cells.

Fig.6. The needed time t for the dispersion of SinGen superlattice with variable periodic length L or atomic number N in the unit cell. The data is obtained by a laptop with an Intel single-core CPU@2.7 THz.
4. Conclusions
In summary, we have proposed a simple and fast tool to calculate the phonon dispersion relation of crystalline solids.When the LAMMPS package is used to simulate crystalline solids,this technique can obtain the interatomic force constant matrix corresponding to the used potential by moving atomic displacements. After obtaining the interatomic force constant matrix,the phonon dispersion relation can be obtained by constructing the dynamic matrix and solving the eigenvalue of the dynamic matrix. Therefore, the proposed technique for the calculation of the phonon dispersion relation can be used to compare and verify the reliability of the potential functions before MD simulation.
We have also used the proposed technique to compare the phonon dispersion relation of graphene with several commonly used potentials. It is found that the Tersoff potential in 2010 and the AIREBO potential can better predict the phonon dispersion relation of graphene than the force field potential,such as REAXFF and PCFF potential. We also use this technique to predict the in-plane and cross-plane phonon dispersions of SinGensuperlattices with much larger unit cells.It is found that there is no phonon band gap along the inplane direction, while there exists a phonon band gap along the cross-plane direction. After obtaining the phonon dispersion relation of the crystal, the sound velocity along the high-symmetry directions of solid and the corresponding elastic constant components can be obtained. In addition, the interatomic force constant matrix would also be used to calculate the thermal conductance from the non-equilibrium Green function technique[30]within the framework of local equilibrium.
猜你喜欢
Chinese Physics B(2022年11期)2022-11-21
Chinese Physics B(2022年5期)2022-05-16
科学与财富(2021年33期)2021-05-10
孩子·小学版(2019年4期)2019-09-10
湘潮(上半月)(2019年12期)2019-05-22
湘潮(上半月)(2019年9期)2019-05-22
海峡姐妹(2016年2期)2016-02-27
校园英语·中旬(2015年6期)2015-07-10
中共党史研究(2013年11期)2013-04-27

- Chinese Physics B的其它文章
- Numerical investigation on threading dislocation bending with InAs/GaAs quantum dots*
- Connes distance of 2D harmonic oscillators in quantum phase space*
- Effect of external electric field on the terahertz transmission characteristics of electrolyte solutions*
- Classical-field description of Bose-Einstein condensation of parallel light in a nonlinear optical cavity*
- Dense coding capacity in correlated noisy channels with weak measurement*
- Probability density and oscillating period of magnetopolaron in parabolic quantum dot in the presence of Rashba effect and temperature*