
Interaction and Mechanism between Imidazolium Ionic Liquids and the Zwitterionic Amino Acid Tyr: a DFT Study
2021-11-22 01:20ZhiweiWuWeiluDingYaqinZhangYanleiWangHongyanHe
物理化学学报 2021年10期
Zhiwei Wu , Weilu Ding , Yaqin Zhang , Yanlei Wang , Hongyan He ,3,*
1 Beijing Key Laboratory of Ionic Liquids Clean Process, Institute of Process Engineering, Chinese Academy of Sciences,Beijing 100190, China.
2 Sino-Danish College, University of Chinese Academy of Sciences, Beijing 100049, China.
3 Zhengzhou Institute of Emerging Industrial Technology, Zhengzhou 450000, China.
Abstract:Ionic liquids (ILs)are thermally and chemically stable and have adjustable structures, which gives them the potential to be used as green, efficient biomolecular solvents. Given the critical role of ILs in dissolving biomolecules, the mechanism of interaction between them deserves further study. Herein, density functional theory (DFT)calculations, using the SMD implicit water solvent model, were employed to study the interaction and mechanism between a hydrophobic zwitterionic amino acid (Tyr)and a series of imidazolium ILs with different alkyl chain lengths and methylation sites. The contributions of hydrogen bonding (H-bonding), electrostatic effects, induction, and dispersion to the intermolecular interactions were determined by combining the symmetryadapted perturbation theory (SAPT), the atoms in molecules (AIM)theory, and reduced density gradient (RDG)analysis.The results indicate that the H-bonding between the IL cation and Tyr is stronger than that between the IL anion and Tyr;however, the binding between either ion and Tyr is dominated by electrostatic effects. By contrast, the difference between the induction and dispersion forces is small when methylation occurs on the C2 site of the imidazolium cation; whereas, it is significantly large when methylation takes place on the N3 site. This is rationalized by the interaction patterns that vary based on the methylation site. H-bonding and π+–π stacking interactions between the imidazole and benzene rings are dominant during C2-methylation, while H-bonding and CAlkyl―H…π interactions between the alkyl chain and benzene ring are dominant during N3-methylation. Increasing the side alkyl chain length has different effects on the interaction energy to cations with different methylation sites. During N3-methylation, when the side alkyl chain length increases from 4 to 12,there are significant van der Waals interactions between the Tyr benzene and the side alkyl chain. However, these van der Waals interactions are inapparent when methylation takes place on the C2 site. Finally, the synergetic effect of the H-bonding and the interaction between the benzene and the side alkyl chain for C2-methylation is greater than the H-bonding and the interaction between the imidazole and benzene rings for N3-methylation, when the side alkyl chain length n > 9.Therefore, the interaction strength and mechanism in these imidazolium-Tyr complexes can be regulated by changing the methylation site and the side alkyl chain length of the cation. Further study of ion-pair and Tyr reveals that the change tendency of the interaction energy of IL-Tyr systems is consistent with that of cation-Tyr cases, and the ion pair further stabilizes the binding with Tyr. These results illustrate the interaction mechanism of IL-Tyr systems and provide a novel strategy for the design and screening of functional ILs for amino acid extraction and separation in the future.
Key Words:Ionic liquids; Zwitterionic amino acid; Interaction and mechanism; Hydrogen bond effect;Van der Waals effect
1 Introduction
Ionic liquids (ILs)are a kind of salts composed of organic cations and organic or inorganic anions, which features with low vapor pressure, excellent chemical stability, and thermal stability1–4.Most of ILs can exist stably in aqueous, acid and alkali environment, and the polarity of solution can be adjusted by tuning the combination of cations and anions to form two-phase or multi-phase separation medium5–7. These unique properties make ILs potential useful solvents for biomolecules8–10. For instance, in ILs, life primary biomolecules such as DNA, amino acid, and protein are found to have good solubility and stability.In particular, hydrophobic biomolecules are more prone to dissolve in the hydrophobic ILs-aqueous two-phase system,whereas difficult in traditional solvents11–13. Generally speaking, the hydrophobicity of ILs has a significant effect on the solubility of biomolecules, which has been illustrated by many studies14–16.
Also, it is found that the cations or anions of ILs with a localized positive or negative charge and the charged amino acid residues of protein molecules can produce evident electrostatic interaction during the protein dissolution process, especially the hydrogen bond (H-bond)widely exists, which plays a vital role in biomolecule solubility17–19. For example, the opening and closing status of Candida Antarctica lipase B (CALB)in ILs 1-butyl-3-methylimidazolium chloride ([Bmim][Cl])and 1-butyl-3-methylimidazolium trifluoromethylsulfonate ([Bmim][TFO])are determined by the H-bonds between amino acid residues and anions20,21. Besides, it has been proved that the large imidazole or pyridyl based cations display apparent van der Waals interactions with amino acids, where the long alkyl chain attached to the cation increases the hydrophobic interaction with biomolecules22,23. In all, the solubility and stability of biomolecules in ILs largely depend on the interactions between ILs and biomolecules, in which electrostatic interaction, H-bonds, van der Waals interaction, and steric effects contribute cooperatively24,25.
Given the critical role of ILs in dissolving and separating biomolecules, it is necessary to address the interaction mechanism deeply15. In this study, the intermolecular interactions between the zwitterion form of hydrophobic amino acid tyrosine (Tyr)and four ILs equipped by series of imidazolium cations which possess different alkyl chain length and methylation site (C4-3mim, C4-2mim, C12-3mim, C12-2mim,see Fig. 1)combined with a hydrophobic tetrafluoroborate anion26(BF4)were studied by DFT calculations. The characterization of various interactions,i.e., H-bonding, induced, and electrostatic interaction, aims at providing how the methylation site and chain length of these considered cations govern the orientation of Tyr in water phase unambiguously. Understanding the effect of the varied chain length and methylation site of ILs on the ILs-amino acid interaction can help to screen and design efficient ILs for dissolving and separating specified biomolecule.
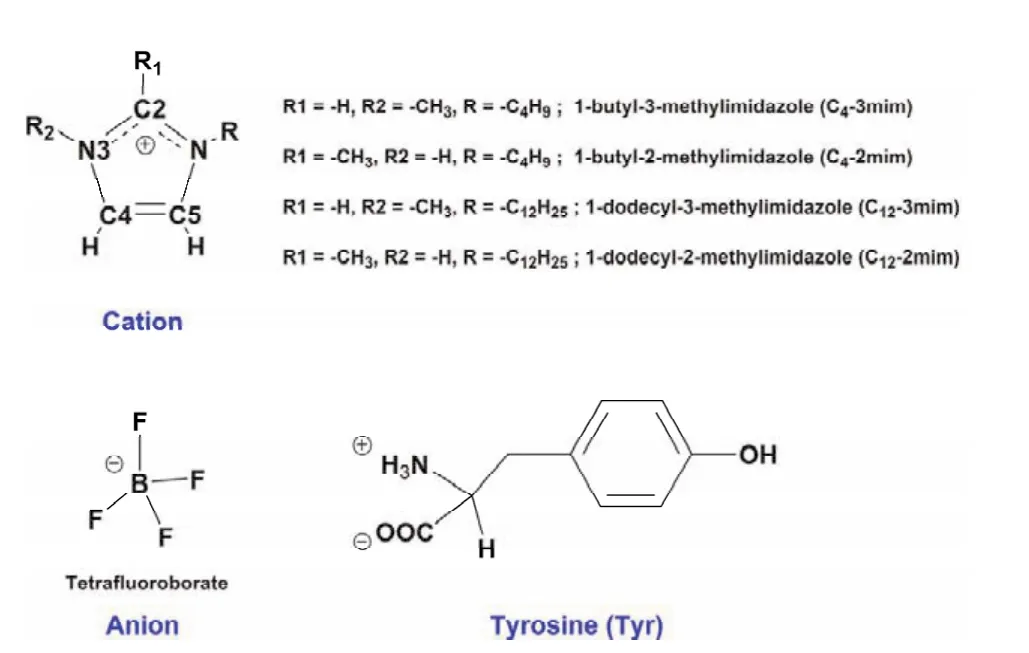
Fig. 1 The structures of studied cations and anion of ILs and amino acid Tyr.
2 Methodology
2.1 Computational details
All DFT calculations were carried out by the Gaussian 09 D.01 program27. For the optimization of isolated cation, anion, and Tyr, the density functional theory (DFT)B3LYP28functional combined with 6-31+G(d,p)basis set were used. As for ILs-Tyr complexes, B3LYP/6-31+G(d,p)and M06-2X29/6-311+G(d,p)level were used for the structure optimization and energy calculation, respectively. The subsequent calculation of frequencies at the same level based on the optimized geometries was performed to determine these geometries being indeed the minima on the potential energy surface. For a better evaluation of dispersion, Grimme’s D3 dispersion correction30was used in all simulation period. Notably, to obtain the reliable structures of cation/anion-Tyr and ILs-Tyr complexes, different inputs as the initial state were constructed, and the most stable one from the energy point was selected to perform the remaining analysis.Simultaneously, to reflect a more realistic environment of the complexes in water solvent, SMD implicit solvent model31,which treats the solution as a continuously polarizable media was used by the structure optimization and energy calculation in Gaussian 09 program32. Further identification of the contribution of electrostatic, exchange, induction, and dispersion items to the total intermolecular interaction energy was analyzed by the symmetry adapted perturbation theory33(SAPT), which was performed in SAPT0/6-311+G(d,p)level as implemented in PSI4 program34. Moreover, the visual analysis of the interaction energy by atoms in molecules (AIM)theory35and the reduced density gradient (RDG)36were completedviaMultiwfn code37.
2.2 Theoretical background
The intermolecular interaction energy was calculated by the following equation:

whereEIon-Tyr(in kJ∙mol−1)is the total energy of cation/anion-Tyr complexes,EIonis the energy of cation or anion, andETyris the energy of Tyr unit in a.u., respectively.
To distinguish the sensitive dependence of many-body effects on complexes binding and accompanying intermolecular interactions, the energy decomposition analysis by SAPT, which gives the contributions from electrostatic, exchange, induction,and dispersion terms to overall binding energy is an efficient tool to quantitative each component38. The electrostatic term (EElst)reflects the electrostatic interaction between molecules, the exchange term (EExch)represents the exchange-repulsion effect,the induction term (EInd)comprises the polarization of a monomer under the other polar molecule as well as the charge transfer between molecules, and the dispersion term (EDis)mirrors the interaction of instantaneous dipoles between nonpolar molecules through the motion of electrons39,40.Accordingly, these four terms can be divided into the attractive forces (EElst,EInd,EDis)and repulsive force (EExch). To provide an intuitive understanding of the proportion of each attractive term to the ΔEIon-Tyr, the contribution percentage for each attractive term can be calculated by the following equation:
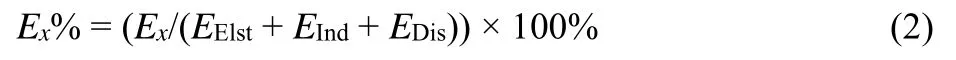
whereEXrepresents any one of the attractive components (EElst,EInd, andEDis).
To identify the strength of the H-bond in its donor-acceptor,the AIM theory41,42can be used to analyze the bonding characteristics qualitatively. There are a set of criteria for the electron density (ρBCP), and Laplacian of the electron density(∇2ρBCP)proposed at bond critical points (BCPs)for the conventional H-bonds43,44. Both parameters for the closed-shell interactions as H-bonds are positive within the ranges of 0.002–0.035 a.u. for theρBCPand 0.024–0.139 a.u. for the ∇2ρBCP.Meanwhile, the Laplacian of electron density at BCPs has positive values indicating that why the nature of these H-bonds is noncovalent. And the negative values of potential energy density (HBCP)at BCPs imply that these H-bonds have the characteristic of covalent bonds, and the positive values of potential energy density (HBCP)indicate these H-bonds have electrostatic properties.
3 Results and discussion
As mentioned above, the ILs are consisted of BF4anion combined with the imidazole cations feature with the different side alkyl chain and methylation site. Therefore, to emphasize the impacts of the side alkyl chain length and methylation site of the cations on the orientation of Tyr in considered ILs-Tyr complexes, the different conformers of isolated cation, anion,and Tyr are optimized to obtain the most stable one in the lowest energy. Then the selected conformer of each species was used to construct the ion-Tyr complexes and do the initial analysis of energy decomposition, AIM, and RDG. Before the discussion, it is noted that the critical atom of each species needs to be defined for clarity: for cation, the ―CH3is substituted on the C2 or N3 site of imidazole ring and the side alkyl chain is substituted on the N1 site, so the H atoms on the C2, N3, and side alkyl chain are labeled as C2―H, N3―H, and CAlkyl―H; for anion, the F atoms are labeled as F1, F2, F3, and F4; as for Tyr amino acid,the N atom is defined as NTyr, the O atoms of ―COO−and ―OH are defined as OCOOand OOH, and the H atoms of Tyr benzene ring and Tyr amino are labeled as CBen―H and CAmino―H,respectively (see Fig. 1).
3.1 ESP and NPA charge analysis
For the aim of predicting the most plausible binding sites aroused by electrophilic and nucleophilic between ion and Tyr complexes, the intuitively surface electrostatic potential (ESP)distributions of cations/anion, and Tyr are firstly investigated.For cation and anion, the most positive region (the blue)generally concentrates on the imidazole ring of each cation, and the most negative region (the red)centers on the BF4anion (Fig.2a). As for Tyr, the ―NH3+group displays positive electrostatic potential, and the ―COO−group shows negative electrostatic potential, which is in accordance with the property of zwitterion and supports the accumulation of ILs on the charged protein surface. Meanwhile, an apparent decrease of electrostatic potential from 625.9 to 538.0 kJ·mol−1can be observed when the―CH3alters from C2 to N3 position (the second and the first panels in Fig. 2b), which significantly reduces the ability of electrophilicity no matter what the side alkyl chain length is. On the other hand, with the extension of the side alkyl chain length,the molecular volume is increased, which is evidenced by the increased surface area with a low electrostatic potential between 70–180 kJ·mol−1(the green shadow labeled in Fig. 2b). Although it has little effect on the high electrostatic surface area distribution of the imidazole ring, a stronger van der Waals potential is still anticipated.
The troll said that he would always be welcome; he had served him faithfully for the three years they had agreed upon, and he could make no objections to his leaving now

Fig. 2 The electrostatic potential of anion, Tyr and cations (a)and the electrostatic potential distributions of cations (b).
To further reflect the strength of the electrophilicity of critical H atom on the imidazole ring (labeled as X―H)quantitatively,the natural population analysis (NPA)is characterized. From Table 1, the variation of the charge of identical X―H in series of cations can be neglected. Meanwhile, the charge of N3―H is almost 2-folds larger than that of C2―H (0.478vs. 0.271), and the charges of C4―H and C5―H (0.266–0.267)are subtly smaller than that of C2―H. The electrophilicity of the H atom of N3―H bond is stronger than that of the C2―H bond.Notably, the charge distribution of CH3―H of C4-3mim and C12-3mim cations significantly reduces by ~51% compared to that of N3―H of C4-2mim and C12-2mim cations, while the distribution of CH3―H of C4-2mim and C12-2mim produces only ~4% decrease with respect to the quantity of C2―H in C4-3mim and C12-3mim. This significant difference is mainly caused by the varied atom type of C and N of H attached. The substitution of―CH3 on the N3 site alters the N―H into C―H. Therefore, the distinct electronic properties of C and N atoms lead to a noticeable decrease in NPA charge of H atom. However, the bond type remains C―H bond when the ―CH3is substituted on the C2 site; thus, the inapparent charge decrease of H atom is observed. These above indicate that regulating the methylation site and side alkyl chain length can affect the electrostatic and van der Waals effect potentially. Through the ESP and NPA analysis, the plausible site of electrophilic for cation and nucleophilic of anion to Tyr can be preliminarily determined.And the construction of cations/anion-Tyr complexes will consider the plausible site.

Table 1 The charge distribution of critical H atom on the imidazole ring in each cation by natural population analysis.
3.2 The characterization of interaction energy of ion-Tyr complexes
3.2.1 The H-bond and interaction energy
Based on the most stable conformer of the isolated ion and Tyr, as well as the plausible site of electrophilic or nucleophilic,the cation-Tyr and anion-Tyr complexes have been constructed.For each complex, several initial configurations are taken into consideration and subjected to the structural relaxation, and the lowest one in total energy is screened out for the analysis of the intermolecular interaction. The most stable conformers have been shown in Fig. 3, and others with their H-bonds properties have been shown in Figs. S1–S3 and Table. S1 (in Supporting Information).
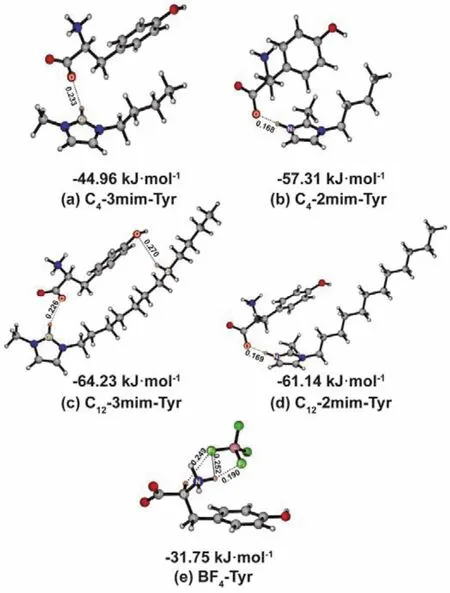
Fig. 3 The most stable conformer and intermolecular interaction energy for cations-Tyr (a–d)and anion-Tyr complexes (e).
Clearly, the benzene ring of Tyr tends to stay around the imidazole ring of C4-2mim and C12-2mim, whereas it orientates toward the side alkyl chain when the ―CH3 of C4-2mim and C12-2mim alters to the N3 site. For the situation of the ―CH3 on the C2 site, the N3―H of cation forms the H-bond with the ―COO−of Tyr where the distances of N3―H…OCOOis ~0.169 nm (see Table 2). After the ―CH3alters to the N3 site, the distances of H-bond, which formed between the C2―H and ―COO−is prolonged by ~0.06 nm significantly. It implies that the change of methylation can determine the strength of H-bond of cation-Tyr complexes because the H-bond involving N3―H is stronger than that of C2―H. On the other hand, the change of the methylation site determines the orientation of Tyr. The methylation on the N3 site makes the stronger N―H H-bond donor change into the weaker C―H H-bond donor, resulting in a similar strengthen of CAlkyl―H compared to C2―H. As aresult, Tyr tends to interact with cation by C2―H…OCOOH-bond and van der Waals effect between benzene ring of Tyr and side alkyl chain of cation collaboratively. As for BF4-Tyr, there are two kinds of F…H H-bonds detected, one is the NTyr―H…F, and another is the CAmino―H…F. Overall, the distances of H-bonds in BF4-Tyr are longer than that in series of cation-Tyr additives, and the angles of H-bonds range from 106°–160°,suggesting that the strength of H-bond in anion-Tyr is weaker than that of cation-Tyr systems.
Apart from the H-bond, the intermolecular interaction energy(ΔEIon-Tyr)of these ion-Tyr complexes further supports the dependence of the stable binding on the methylation site and side alkyl chain qualitatively. For the cations feature with shorter side alkyl chain (C4-2mim and C4-3mim), the ΔEIon-Tyr are −57.31 and−44.96 kJ·mol−1, which are smaller than the values of longer side alkyl chain constructed C12-2mim and C12-3mim (−61.14 and−64.23 kJ·mol−1). The ΔEIon-Tyrof C4-2mim-Tyr is obviously stabilized by 12.35 kJ·mol−1than that of C4-3mim-Tyr and the difference in ΔEIon-Tyrof two long side alkyl chain based cation-Tyr cases is only 3.09 kJ·mol−1. It is worth noting that with the increase of side alkyl chain length, the two systems show an opposite trend of interaction energy, which is C4-2mim-Tyr

Fig. 4 The interaction energy of cation-Tyr complexes with alkyl chain length growth.
3.2.2 Energy decomposition analysis
After analyzing the interaction energy of ion-Tyr complexes,we will turn the concentration to the proportions from the different attractive components to the ΔEIon-Tyr, containing electrostatic (EElst), induction (EInd)and dispersion (EDis)terms as depicted in the theoretical background. The contributions of these attractive components in each ion-Tyr case are illustrated in the form of a histogram in Fig. 5.

Fig. 5 The contribution percentage of the SAPT-derived constructive components of cation-Tyr and anion-Tyr systems.
For a series of cation-Tyr systems, the dominant factor to the binding comes from theEElst term that it contributes by one half to the total interaction energy in each case. However, the percentage of theEDisandEIndterms shows a different change tendency from case to case. For ―CH3substituted on the N3 site, theEDisis more obvious than that on the C2 site, which is mainly attributed to the fact that the C2―H…CCOOH-bond makes Tyr shifting to the side alkyl chain of cation, subsequently induces the CAlkyl―H…πinteraction45between the side alkyl chain and non-polar benzene ring of Tyr. Oppositely, theEIndis overweight than theEDiswhen the ―CH3locates on the C2 site,and the main reason is that the N3―H…CCOOH-bond attracts Tyr keeping away from the side alkyl chain, consequently produces theπ+–πinteraction46between the polar imidazole ring and non-polar benzene ring. It is worth noting that the difference betweenEDisandEIndin C4-2mim-Tyr and C12-2mim-Tyr is smaller than that in C4-3mim-Tyr and C12-3mim-Tyr, accounting that extending the side alkyl chain of the cation is less effective in the formation of the CAlkyl―H…πinteraction when ―CH3situates on the C2 site of cation; therefore, the H-bonding effect dominates the whole interaction.
Differently, in C4-3mim-Tyr and C12-3mim-Tyr, the C2―H…OCOOH-bond makes Tyr oriented to the side alkyl chain of cation, consequently produces the CAlkyl―H…πinteraction as the extending of the side alkyl chain. Therefore,the electrostatic and van der Waals effect collaborate to the binding of cation and Tyr. As for BF4-Tyr complexes, it can be found that theEElstis overwhelmingly larger than theEDisandEInd, and the contributions from the latter two terms are relatively lower in less than 10% and 20%. At the same time, theEElstof anion-Tyr is significantly larger by ~20% than that of cation-Tyr cases, illustrating why the electrostatic effect nearly governs the binding of BF4 and Tyr completely, while the van der Waals effect participates more in combining of cation and Tyr.
3.2.3 AIM analysis
Although H-bond distance and angle can speculate the H-bond strength qualitatively, the more details associated with the electron density of H-bond are still not obtained. According to the AIM theory, the electron density (ρBCP), Laplacian of the electron density (∇2ρBCP)and the potential energy density (HBCP)can be used to illustrate the strength and nature of H-bond at the bond critical points (BCPs)qualitatively. The H-bond and ionic bond are identified by ∇2ρBCP> 0 in the closed-shell interactions,while the covalent bond is characterized by ∇2ρBCP< 0; the extremely strong H-bond can also have negative Laplacian value; and the positive value of HBCPindicates the H-bond has electrostatic properties, separately.
The magnitudes ofρBCP, ∇2ρBCP, andHBCPof all considered ion-Tyr complexes have been summarized in Table 3. Totally,theρBCPand ∇2ρBCPof most H-bonds in considered complexes fall in the relative proposed ranges (0.002–0.035 a.u. for theρBCP and 0.024–0.139 a.u. for the ∇2ρBCP)where the former ranges in 0.007–0.047 a.u. and the latter ranges in 0.021–0.138 a.u. in our study, respectively. For the H-bond of the series of cation-Tyr complexes, the strength of H-bond decreases by order of C4-2mim-Tyr (0.047 a.u.)≈ C12-2mim-Tyr (0.045 a.u.)> C12-3mim-Tyr (0.014 and 0.007 a.u.)> C4-3mim-Tyr (0.011 a.u.),respectively. It is worth noting that ―CH3substituted on the C2 site of imidazole ring induces more strengthened H-bond(N3―H…OCOO)with the largestρBCPand ∇2ρBCPby 0.047 a.u.and 0.137 a.u. separately with respect to the situation of on the N3 site (the ∇2ρBCPis 0.039 and 0.047 a.u. for C4-3mim-Tyr and C12-3mim-Tyr). Simultaneously, ―CH3 located on the C2 site produces a negative value ofHBCP(−5.634 × 10−3and −4.675 ×10−3a.u.), indicating that methylation on this site features astrong electrostatic effect compared to that on the N3 site. These above mentioned again confirm that methylation on the C2 site is more favorable than that on the N3 site in the formation of stronger H-bond and further stabilization of the binding. As for BF4-Tyr system, the strong NTyr―H…F2 (0.023 a.u.)and weak CAmino―H…F1 (0.008 a.u.)can be detected, while the electron density at NTyr―H…F1 is undetectable which is mainly aroused from the relatively longer H-bond distance (0.252 nm)and smaller H-bond angle (106.9°)compared to others.

Table 3 The electron density (ρBCP)and the associated value of Laplacian (∇2ρBCP), as well as the potential energy density (HBCP)at the bond critical point in cation-Tyr and anion-Tyr complexes.
In all, the methylation site change between the C2 and N3 site on the imidazole ring determines the C2―H…OCOOand N3―H…OCOOH-bond strength of cation-Tyr systems because the N3―H donor manifests stronger than the C2―H donor.Compared to the distinct influence of varying the methylation site on the strength of H-bond, the impact of extending side alkyl chain is faint. Oppositely, the H-bond strength in BF4-Tyr complexes is moderate with respect to that in C4-2mim-Tyr and C12-2mim-Tyr, suggesting the critical role of appropriate methylation site of the imidazole-based cation in interacting with Tyr preferentially.
3.2.4 RDG analysis
Apart from the useful tool of AIM to identify the nature of noncovalent, reduced density gradient (RDG)analysis is another powerful tool to explore noncovalent interaction intuitively. This method can provide more conspicuous information compared to AIM analysis in a deep comprehension of the varied interaction mechanism to noncovalent interaction. RDGvs. the electron density multiplied by the sign of second Hessian eigenvalue(sign(λ2)ρ)is plotted by scatter graph (see the left panel in Fig.6a–e)where the more negative value represents stronger electrostatic/H-bonding interaction (the blue), the positive value represents steric effect (the red), and the region close to zero represents van der Waals effect (the green), respectively.
In the series of cation-Tyr complexes, the location of the blue spikes in C4-2mim-Tyr and C12-2mim-Tyr falls into the more negative region (maximum to −0.047 a.u.)compared to C4-3mim-Tyr and C12-3mim-Tyr (maximum to −0.014 a.u.).Moreover, the corresponding region color of the disc-shaped blocks (the labeled circle in the right panel in Fig. 6a–e)for the N3―H…OCOOin C4-2mim-Tyr and C12-2mim-Tyr is dark blue in contrast to the green for the C2―H…OCOOin C4-3mim-Tyr and C12-3mim-Tyr. It is noted that in C4-2mim-Tyr and C12-2mim-Tyr, the van der Waals interaction is observed between the benzene ring of Tyr and the imidazole ring of cation, while it is subtle between the benzene ring and the side alkyl chain,assuring that extending the side alkyl chain of cation produces little effect on the binding when methylation situates on the C2 site. Conversely, for C4-3mim-Tyr and C12-3mim-Tyr, the benzene ring orientates to the side alkyl chain due to the formation of C2―H…OCOOthus increases the CAlkyl―H…πinteraction. These combined directly imply that both of C4-2mim-Tyr and C12-2mim-Tyr display a stronger H-bonding effect andπ+–πinteraction compared to C4-3mim-Tyr and C12-3mim-Tyr which manifests a weaker H-bonding effect and CAlkyl―H…πinteraction, assuring that extending the side alkyl chain induces little effect on the van der Waal interaction between the benzene ring of Tyr and side alkyl chain of cation when methylation situates on the C2 site, whereas boosts the significant van der Waals interaction when methylation locates on the N3 site, separately. As for BF4-Tyr complexes, the location of the blue spikes falls into the more positive region(−0.023 a.u.)with respect to that of C4-2mim-Tyr and C12-2mim-Tyr complexes. And the corresponding region color of the discshaped blocks for NTyr―H…F H-bond is green, suggesting an obvious NTyr―H…σinteraction between the benzene ring and BF4on the other hand. These above reflect that C4-2mim and C12-2mim can interact with Tyr more easily, follows by the BF4anion as well as C4-3mim and C12-3mim. It is good in line with the H-bond distances and the AIM analysis, and considering the strong H-bonding effect among the C4-2mim-Tyr, C12-2mim-Tyr, and BF4-Tyr, it can be anticipated that their combinations,namely [C4-2mim][BF4] and [C12-2mim][BF4], binding to Tyr can behave an excellent performance in extracting and separating Tyr.
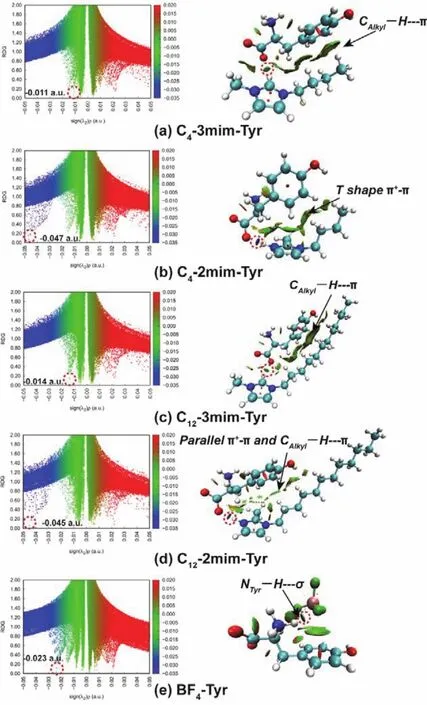
Fig. 6 The RDG scatter plot and surface plot of each cation-Tyr and anion-Tyr complexes (a–e).
3.3 The interaction of IL and Tyr
Because cation and anion appear in the form of ion pair in the real ILs which behaves more complicated structure, therefore, to obtain the interaction and mechanism between ILs and Tyr more appropriately, it is necessary to explore the structural behavior of ILs-Tyr complexes which is essential to reveal the extracting and separating mechanism. Based on the considered cations and anion, four kinds of ILs are constructed, namely [C4-2mim][BF4], [C4-3mim][BF4], [C12-2mim][BF4], and [C12-3mim][BF4], respectively. The varied initial conformations and H-bonds properties of each IL-Tyr complexes have been shown in Fig. S6 and Table S2 (in Supporting Information), and the optimized most stable one has been shown in Fig. 7.

Fig. 7 The optimized conformers and intermolecular interaction energy for IL-Tyr complexes.
The evaluation of the interaction between IL and Tyr is calculated by the following equation47:

where theEIL-Tyr(in kJ·mol−1)is the energy of IL-Tyr complexes,EILandETyrare the energies of isolated IL and Tyr calculated on the structure of IL-Tyr complexes, respectively. As seen from Fig. 7, zwitterion Tyr has an electron-withdrawing group―COO−and an electron-donating group ―NH3+, which acts as a bridge for connection of cation and anion. All kinds of ion can interact with TyrviaH-bond where anion locates around theand cation situates toward the ―COO−. The orientation of cation and anion in each stable IL-Tyr complex is similar to that in the standalone ion-Tyr complexes.
The ΔEIL-Tyr of IL-Tyr is more stabilized than that of the single cation-Tyr or anion-Tyr system that it decreases from [C12-3mim][BF4] (−95.61 kJ·mol−1)> [C12-2mim][BF4] (−88.73 kJ·mol−1)> [C4-2mim][BF4] (−81.90 kJ·mol−1)> [C4-3mim][BF4] (−66.70 kJ·mol−1), respectively. This is in accordance with the change tendency of ΔEIon-Tyrof cation-Tyr complexes listed in Table 2, suggesting that introducing anion to the cation-Tyr system does not break the binding between cation and Tyr. To further identify the difference of interaction mechanism between ion-Tyr and IL-Tyr complexes, the varied contributions from H-bond and van der Waal effect are analyzed in Table 4 and Fig. 8. From Table 4, it can be found that the strength of H-bond in [C4-2mim][BF4]-Tyr and [C12-2mim][BF4]-Tyr is stronger than that of [C4-3mim][BF4]-Tyr and [C12-3mim][BF4]-Tyr that theρBCPranges from 0.007–0.050 a.u. for the former two and 0.011–0.022 a.u. for the latter two.Meanwhile, the value ofHBCPin [C4-2mim][BF4]-Tyr (−7.241 ×10−3– 1.922 × 10−3a.u.)and [C12-2mim][BF4]-Tyr (−5.678 ×10−3– 1.959 × 10−3a.u.)is more negative compared to that in[C4-3mim][BF4]-Tyr (1.300 × 10−3− 2.007 × 10−3a.u.)and [C12-3mim][BF4]-Tyr (0.556 × 10−3–2.022 × 10−3a.u.), implying the H-bond in the latter two systems possess more obvious electrostatic effect.
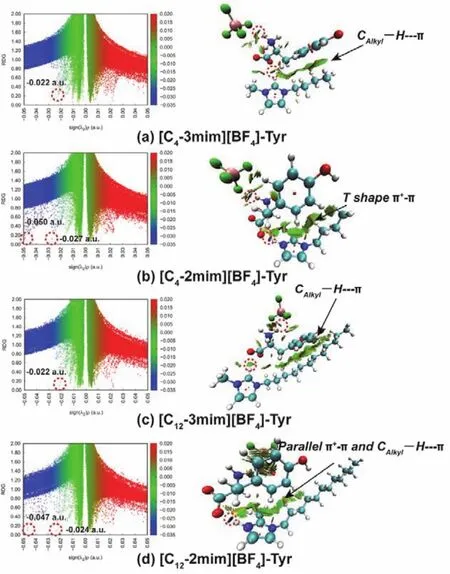
Fig. 8 RDG scatter plot and surface plot of each IL-Tyr complexes.

Table 4 The electron density (ρBCP)and the associated value of Laplacian (∇2ρBCP), as well as the potential energy density (HBCP)at the bond critical point in ILs-Tyr complexes.
As a whole, H-bond, electrostatic, and van der Waals effects have been observed in these IL-Tyr systems; their synergistic effects help to stabilize the binding between IL and Tyr. This insight accounts for the different extents of our considered ILs in extracting and separating Tyr amino acid. We believe that this can provide a novel strategy in design functional ILs in extracting and separating zwitterion amino acid in the future.
4 Conclusions
In summary, the intermolecular interaction and mechanism between a series of [cation][BF4] ILs and zwitterion amino acid Tyr have been investigated by DFT calculations in this work. For the series of the cation (C4-2mim, C4-3mim, C12-2mim, and C12-3mim), they possess a varied length of side alkyl chain and methylation site.Viacharacterizing the strengthen of H-bond,ESP, NPA, AIM, and RDG analysis of isolated cation-Tyr and anion-Tyr systems, it can be found that the interaction between each cation-Tyr is stronger than that of BF4-Tyr. And the methylation on the N3 site makes the strong N―H H-bond donor change to weak C―H H-bond donor, and H-bond alters from N3―H…OCOOin C4-2mim-Tyr and C12-2mim-Tyr to C2―H…OCOOin C4-3mim-Tyr and C12-3mim-Tyr. The methylation on the C2 site adjusts Tyr binding to strong N3―H H-bond donor. Meanwhile, it further obtains from the energy decomposition analysis that electrostatic term mainly dominates the binding between all kinds of ion and Tyr, and the difference between the induction and dispersion terms in C4-2mim-Tyr and C12-2mim-Tyr is smaller than that in C4-3mim-Tyr and C12-3mim-Tyr, confirming that extending side alkyl chain is less effective in reinforcement the binding because the methylation on the C2 site of cation hinders the benzene ring-alkyl chain interaction. Conversely, this difference is more apparent in C4-3mim-Tyr and C12-3mim-Tyr cases, which stems from the fact that the Tyr is oriented to the side alkyl chain when methylation on the N3 site, as a result, forming a weaker C―H H-bond donor of the cation. Consequently, the binding of C4-2mim and C12-2mim with Tyr is mainly induced by the H-bond,π+–πstacking interaction, and in C4-3mim and C12-3mim related cases, it is mainly governed by the H-bond and CAlkyl―H…πinteraction,respectively. Whereas for BF4-Tyr, the interaction is smaller than cation-Tyr systems, and the H-bond of NTyr―H…F is the governed item to the binding of BF4and Tyr. The results of the complexes between ion pair and Tyr show that the change tendency of interaction energy of IL-Tyr systems is in line with that in cation-Tyr cases, and the ion pair further stabilizes the binding with Tyr. And the interaction strength and mechanism in cation-Tyr complexes can be adjusted by changing the methylation site and side alkyl chain length of the cation.Meanwhile, the anion further stabilizes the IL-Tyr complexes collaboratively. These findings potentially help to screen and design of biocompatible ILs in extracting and separating zwitterion amino acid in the future.
Supporting Information:available free of chargeviathe internet at http://www.whxb.pku.edu.cn.
Acknowledgment:Sincere thanks to Prof. Suojiang Zhang(IPE, CAS)for his guidance and help.

- 物理化学学报的其它文章
- Hollow Nitrogen-Rich Carbon Nanoworms with High Activity for Metal-Free Selective Aerobic Oxidation of Benzyl Alcohol
- Photocrosslinking-Immobilized Polymer Vesicles for Lowering Temperature Triggered Drug Release
- CO Hydrogenation to Ethanol over Supported Rh-Based Catalyst:Effect of the Support
- CdTeSe合金幻数团簇的室温合成和形成机理研究
- 体相界面导通的复合快离子导体
- 异氰酸苯酯诱导的类胶原多肽自组装