
Atomistic Insight into Changes in the Vibrational Spectrum of Ionic Liquids under External Electric Field
2021-11-22 01:20WenqiongChenYongjiGuanJiaoZhangJunjiePeiXiaopingZhangYouquanDeng
物理化学学报 2021年10期
Wenqiong Chen , Yongji Guan , Jiao Zhang , Junjie Pei , Xiaoping Zhang ,*, Youquan Deng
1 Institute of Optoelectronics and Electromagnetic Information, School of Information Science and Engineering, Lanzhou University,Lanzhou 730000, China.
2 Centre for Green Chemistry and Catalysis, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000,China.
Abstract:Vibrational spectroscopy is a powerful tool for studying the microstructure of liquids, and anatomizing the nature of the vibrational spectrum (VS)is promising for investigating changes in the properties of liquid structures under external conditions. In this study,molecular dynamics (MD)simulations have been performed to explore changes in the VS of 1-ethyl-3-methylimidazolium hexafluorophosphate ([Emim][PF6])ionic liquid (IL)under an external electric field (EEF)ranging from 0 to 10 V∙nm−1 at 350 K. First, the vibrational spectra for [Emim][PF6] IL as well as its cation and anion are separately obtained, and the peaks are strictly assigned. The results demonstrate that the VS calculated by MD simulation can well reproduce the main characteristic peaks in the experimentally measured spectrum. Then, the vibrational spectra of the IL under various EEFs from 0 to 10 V∙nm−1 are investigated, and the intrinsic origin of the changes in the vibrational bands (VBs)at 50, 183, 3196, and 3396 cm−1 is analyzed. Our simulation results indicate that the intensities of the VBs at 50 and 183 cm−1 are enhanced. In addition, the VB at 50 cm−1 is redshifted by about 16 cm−1 as the EEF is varied from 0 to 2 V∙nm−1, and the redshift wavenumber increases to 33 cm−1 as the EEF is increased to 3 V∙nm−1 and beyond. However, the intensities of the VBs at 3196 and 3396 cm−1 show an obvious decrease.Meanwhile, the VB at 3396 cm−1 is redshifted by about 16 cm−1 when the EEF increases to 3 V∙nm−1, and the redshift increases to 33 cm−1 with an increase in the EEF beyond 4 V∙nm−1. The intensity of the VB at 50 cm−1 increases because of the increase in the total dipole moment of each anion and cation (from 4.34 to 5.46 D), and the redshift is attributed to the decrease in the average interaction energy per ion pair (from −378.7 to −298.0 kJ∙mol−1)with increasing EEF. The intensity of the VB at 183 cm−1 increases on account of the more consistent orientations for cations in the system with increasing EEF. The VB at 3196 cm−1 weakens visibly because a greater number of hydrogen atoms appear around the carbon atoms on the methyl/ethyl side chains and the vibrations of the corresponding carbon-hydrogen bonds are suppressed under the action of the EEF. Furthermore, the intensity of the VB at 3396 cm−1 decreases due to the decrease in the intermolecular +C―H··F− hydrogen bonds (HBs), while the relaxation effect that is beneficial for the formation of HBs simultaneously exists in the system under the varying EEF, thus causing a redshift of the VB at 3396 cm−1.
Key Words:External electric field; Vibrational spectrum; Ionic liquids; Molecular dynamics simulation
1 Introduction
Room temperature ionic liquids (RTILs)are substances generally composed of specific anions and cations and have presented widespread applications in chemical industry field due to their excellent physicochemical properties1–5. Previous research work has indicated that the spectroscopic method is useful to characterize molecular structures in ionic liquid (IL)materials including hydrogen bonds (HBs)and various conformational groups6–8. The vibrational spectra of some imidazole ILs have already been studied by using experimental and computational methods, which investigates the probably existing and stable molecular structures, the formation of HBs and the interaction energies between anions and cations in ILs9–17.Bhargava and Balasubramanian10calculated the vibrational spectrum (VS)of 1,3-dimethylimidazolium chloride IL([Mmim][Cl])through the Fourier transform of the normalized velocity auto-correlation function (VACF)for all atoms in the system based on Car-Parrinello molecular dynamics (MD)simulation method. It was found that this calculation result is basically consistent with the experimental spectral data and shows that the HBs formed by chloride anions and acidic hydrogen atoms on the cationic imidazole rings could be acquired by both structural data and a more direct method of cationic vibration dynamics. Afterwards the stable and possible variants of ion pairs molecular structures that were determined with the anion positioned in several common imidazole ILs were explored by using the combined density functional theory(DFT)-vibrational spectroscopy approach and it revealed that both conformational change and counter ions decide the cations vibrations11. In 2008, Jeon and co-workers12employed the attenuated total reflection infrared absorption spectra to investigate structures of 1-butyl-3-methylimidazolium iodide and tetrafluoroborate ([Bmim]I and [Bmim][BF4])ILs and their aqueous mixtures. The experimental results showed that the vibrational spectra of [Bmim]+cations in these two ILs are very unlike and change with the water concentration increase, which were caused by the different relative positions between the anions and cations. Moreover, the first Fourier transform infrared spectroscopy (FTIR)measurements of several imidazole ILs with 1-ethyl-3-methylimidazolium [Emim]+cation and thiocyanate ([SCN]−), dicyanamide ([N(CN)2]−),ethyl sulfate ([EtSO4]−)and bis(trifluoromethylsulfonyl)imide([NTf2]−)anions, respectively, have been presented in the far infrared spectral region16and it offered a direct probe to study the strength of interaction energies between anions and cations in ILs. Ludwiget al.17also investigated the influence of HBs on the ILs physical properties by using a suitable combination of experimental and theoretical method, and confirmed that HBs have a significant influence on the melting points, viscosities and enthalpies of vaporization for ILs through far-infrared spectroscopy and DFT calculations. Thomaset al.6firstly presented anab initiomolecular dynamics analysis of vibrational spectra of ILs consisting of ([Emim]+)cation and acetate anion and its mixtures with water and carbon dioxide.Through comparing the results with experimental data, they confirmed that the applied computational method is suited to study the influence of complicated bulk phase system model on vibrational spectra in systems, which were very important for discussing the microscopic structure of the system having a dynamics network with strong intermolecular interactions.Brelaet al.18performed extensive analysis of the HB network and its effect on vibrational spectra in a typical aprotic IL (1-ethyl-3-methylimidazolium bis-(trifluoromethylsulfonyl)imide([Emim][TFSI])by usingab initioand classical MD simulations.Furthermore, the molecular interactions of a Cu-based metalorganic framework with a confined imidazolium-based IL were investigated through vibrational spectroscopy study by using the method combined DFT and experiment19, and the solvation of AgTFSI in [Emim][TFSI] IL was investigated by vibrational spectra and DFT calculations20. Recently, the vibrational spectra of ring hydrogen atoms of imidazolium-based ILs with [Bmim]+cations paired with Cl−, Br−and [BF4]−anions were studied by using classical MD simulations with different force fields and DFT methods using the MD results for liquid structures in the QM/MM (quantum mechanics/molecular mechanics)framework21. The results show that the C―H stretching vibrations captured with CHARMM force field present a large departure from experiments, while the OPLS-AA force field leads to a similar deviation but to a lesser degree. On the other hand, the QM/MM results correctly predict that C―H vibrations shift to lower frequencies as the basicity of the HB accepting anions increases, in good accordance with experiments. In addition, Paschoalet al.have reviewed the conceptual basis of theoretical frameworks which have been used to interpret vibrational spectra of ILs22. The issues addressed in this review are the intermolecular vibrations that are more directly probed by the low-frequency range of IR spectra and the applications of vibrational spectroscopy in studying phase transitions of ILs.
As is known to all, electric fields are of great importance for studying the microscopic and macroscopic regulation characteristics of IL materials. The structural properties of ILs under the external electric field (EEF)and electric field actuation of IL polymer have attracted a lot of attention of researchers in recent years23–28. In 2009, Wang27revealed the phenomenon of a disordering and reordering procedure of the change in the ILs structure using non-equilibrium MD simulations, and found that the IL structure disorders from spatially heterogeneous to homogeneous and then reorders to nematic-like with the EEF strength increase. Around the same time, Daily and co-worker24estimated electrical conductivity and ionic velocities in[Emim][BF4] IL at the high EEFs ranging from 4 × 108to 4 ×1010V·m−1using MD simulations, and they perceived that the predictive electric conductivities keep growing with electrical field until that thermostat effect becomes important. The VS is one of the most powerful analytical tools to exhibit molecular structures, while there are few reports about the spectral characteristics of ILs under the different EEFs. The calculation of the VS using MD simulation method can obtain the spectral characteristic of the larger system and more comprehensively analyze the interactions between anions and cations and exclude the influence of impurities on the IL spectrum measurement with respect to the DFT calculations and experimental measurements.In our previous research work29, we preliminarily calculated and assigned the VS of 1-ethyl-3-methylimidazolium hexafluorophosphate ([Emim][PF6])imidazole IL in a bulk system using MD simulation in the range from 0 to 4000 cm−1,and qualitatively analyzed the variations of intensities and positions of the VS with 3 V·nm−1EEF, but the intrinsic origin of the changes for vibrational bands (VBs)under different electric fields is still unclear.
Herein, to further probe the intrinsic origin of the electric field induced changes for VBs, the MD simulation is employed to separately calculate the vibrational spectra for the cation and anion of [Emim][PF6] IL under 0 V·nm−1EEFviathe Fourier transform of normalized VACFs and the VBs are strictly assigned according to the DFT calculation and experimental results in the literature11,13. Meanwhile, the calculated results of the VBs of the [Emim][PF6] IL at 50, 183, 3196 and 3396 cm−1with a series of varied EEFs ranging from 0 to 10 V·nm−1reveal that the intensity and position of the VS indeed change by the varied EEFs. We make a further exploration for the origin of the changes in combination with the data from MD simulations,which are mainly attributed to the strengthening of the total dipole moment for per anion and cation and weakening of the average interaction energy for per ion pair under the EEF action.Thus the EEF makes the orientations of cations more consistent and increases the number of hydrogen atoms around the carbon atoms on the methyl/ethyl side chains, and furthermore breaks the existing intermolecular+C―H··F−HBs in the system but emerges a relaxation effect that is beneficial to the formation of HBs at the same time.
2 Simulation details
In this work, the vibrational spectra of imidazole IL[Emim][PF6] under different EEFs ranging from 0 to 10 V·nm−1are performed by using MD simulations, and the geometrical structures of the anion and cation are shown in Fig. 1a. A bulk[Emim][PF6] IL system, comprised of 512 pairs of [Emim]+cations and [PF6]−anions, was firstly arranged within a 5.16 nm ×5.16 nm × 5.16 nm cubic simulation cell constructed by using PACKMOL software30. The structure displayed in Fig. 1b is the initial structure of ensemble with the cube model resulting from a single MD simulation, which can be visualized by using the visual molecular dynamics (VMD)graphics software31. The periodic boundary conditions are used in each direction of the simulation box during the entire MD simulation process.
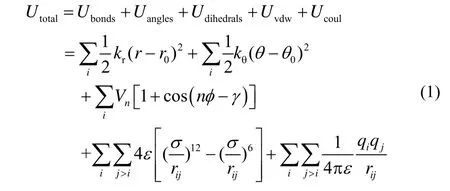

Fig. 1 Geometry structures of the 1-ethyl-3-methylimidazolium cation ([Emim]+)and hexafluorophosphate anion ([PF6]−)(a)and initial structure of the ensemble including cubic bulk ILs (b).
In this work, all MD simulations are performed through the general purpose parallel MD simulation open-source package DL_POLY 4.0832. The polarizable force field developed by Wanget al.22is used for [Emim]+and the systematic all-atom force field (AA-FFs)developed by Canongia Lopeset al.33that is in the frame of optimized potential for liquid simulation/all atom (OPLS-AA)force field34,35frame structure is used for[PF6]−. The potential energy of the system is simulated by the following standard functional expression:where the first three terms stand for the intramolecular bond stretching, angle bending, and dihedral torsion interactions, and the last two terms describe the intermolecular non-bonded interactions. The van der Waals parameters of the interactions between different atoms are obtained from the Lorentz-Berthelot combining rules. The electrostatic Long-range interactions are calculated by using the Ewald summation method36with a precision of 1 × 10−6. The 1.5 nm cutoff distance is taken for the vdW and electrostatic interactions. The Newton’s equations of motion are integrated by using a velocity verlet algorithm with 2.0 fs integration time step. It randomly assigns the initial velocities to all atoms in the system in accordance with the temperature of the system. The simulation is performed with the Nosé-Hoover thermostat/barostat algorithm37,38and the temperature of the thermostat is set based on the melting point of [Emim][PF6] IL, where the initial MD simulation is performed with the NPT ensemble forT= 350 K andp= 1.01325 × 105Pa until the system’s total energy converges to a constant value after 10 ns. Then a NVT ensemble is run 10 ns again in order to maintain a better equilibrium condition, where the starting point of the NVT ensemble is the equilibrated configuration of the NPT ensemble atT= 350 K. Finally, after reaching the equilibrium state, a series of different EEFs ranging from 0 to 10 V·nm−1carried out only in the y direction are separately applied on the bulk IL [Emim][PF6] configuration to collect statistically equilibrium averaged values taking per 1000 time steps in the following 10 ns.
3 Results and discussion
3.1 VS with 0 V·nm−1 EEF
Vibrational spectroscopy is a universal tool for studying ILs molecular structures and HBs under different external conditions in recent years. In this work, the spectra are obtained from MD simulations through the Fourier transform of the VACFs of[Emim][PF6] IL in the system. The normalized VACF in timetis defined as39

wherevi(t)is the velocity of atomiof the [Emim][PF6] IL at timetand the angular brackets denote the ensemble average. The normalized VACFs of all atoms show rapid convergence in a small vibration period of about 2 ps within the strong interaction molecular system, including a large number of atoms and molecules. The VS can be calculated through the Fourier transform of equivalent normalized VACFs for all atoms in the system10,40
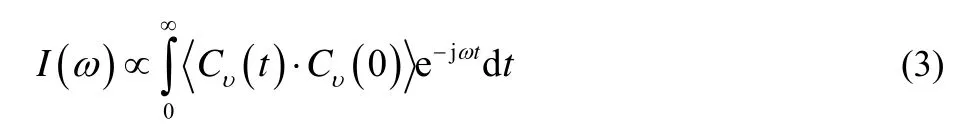
whereI(ω)represents the intensity of the calculated spectrum,Cυ(t)is the centroid equivalent normalized VACFs for corresponding atoms of the simulation system in timet,ωis the spectral wavenumbers, and the angular bracket represents the mean value of all the time origins. The [PF6]−VS with the range between 10 and 1000 cm−1, and [Emim]+and [Emim][PF6] IL vibrational spectra in the range of 10 to 4000 cm−1with 0 V·nm−1EEF are obtained from the calculation method in Fig. 2,respectively. We strictly assign the VBs through comparing the spectra obtained from DFT calculation and experimental measurements11,16,41in Table S1 (in Supporting Information). As mentioned in our previous work29, the calculated VS of IL using MD simulation can reproduce the main features of the experimentally measured spectra very well. The bands below 100 cm−1are assigned as the bending and stretching modes of cation-anion, which can be a sensitive probe for the average interaction energy between anions and cations16,42. The 183 cm−1can be assigned as the torsional vibration band of the ethyl side chain. The 266 cm−1and 632 cm−1are attributed to the out of plane bending band of the CH3―(N)methyl group and the CH3CH2―(N)ethyl group on the cationic imidazole ring. The band at 416 cm−1is assigned to the bending modes of the anions,while the 583, 783 and 949 cm−1are the stretching bands of the anions. The 3196 cm−1band is attributed to the anti- and symmetrical stretching vibration modes of C―H bonds on methyl and ethyl side chains, and the 3396 cm−1band is assigned to the C2―H2, C4―H4 and C5―H5 stretching vibration band.The other bands between 400 and 4000 cm−1are attributed to the stretching, bending, rocking and twisting of the groups and bonds on the cationic imidazole ring and methyl and ethyl side chains.

Fig. 2 The vibrational spectra (arbitrary units)of (a)[Emim]+ cation, (b)[PF6]− anion and (c)[Emim][PF6] IL calculated by Fourier transforming of all atoms’ VACFs with 0 V·nm−1 EEF.
3.2 Vibrational spectra with varied EEFs
To study the effect of different EEFs on the VS of[Emim][PF6] IL, the vibrational spectra are investigated by MD simulation methods carried out with EEFs varying from 0 to 10 V·nm−1with interval of 1 V·nm−1. As the EEF gradually increases, the intensities and positions change of the vibrational spectra are presented in Fig. S1 (in Supporting Information). By comparison, we can find that the intensities and positions of VBs below 200 cm−1or above 3000 cm−1are significantly changed with the varied EEFs. In order to better analyze the change of corresponding vibrational peaks, we presented the VBs at 50 and 183 cm−1in Fig. 3a and at 3196 and 3396 cm−1in Fig. 3b,respectively. We can see from Fig. 3a that the 50 cm−1band shifts to a low frequency by 16 cm−1(redshift)with 2 V·nm−1EEF and the redshift wavenumbers increase to 33 cm−1with EEF exceeding 3 V·nm-1, which are accompanied by a continuous enhancement in the intensity of the band, while the intensity of the 183 cm−1band slightly increases with the varied EEFs increasing. The phenomena can be observed in Fig. 3b that the intensities of VBs at 3196 and 3396 cm−1are obviously weakened as the EEFs increase from 0 to 10 V·nm−1, meanwhile the 3396 cm−1band redshifts 16 cm−1with 3 V·nm−1EEF and shift wavenumbers increase to 33 cm−1as the EEF beyond 4 V·nm−1.
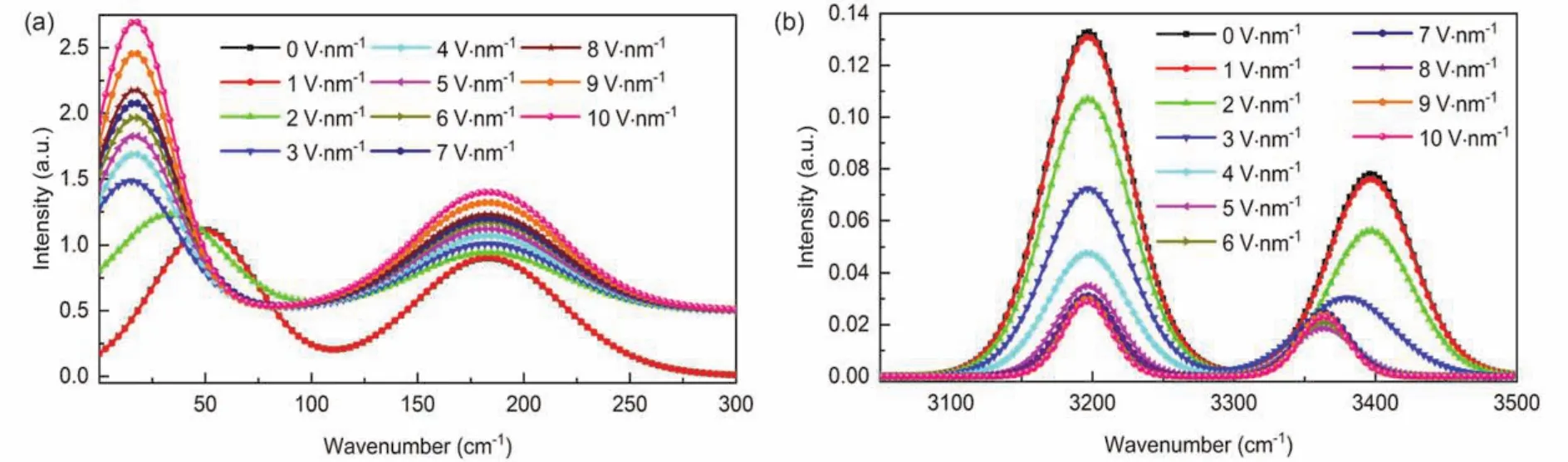
Fig. 3 The vibrational spectra (arbitrary units)for [Emim][PF6] IL with the varied EEFs in the range of 0 to 10 V·nm−1 for(a)the spectral range is between 10 and 300 cm−1 and (b)the spectral range is between 3050 and 3500 cm−1.
In the case of 50 cm−1VB, the intensity continues to enhance with the increase of the varied EEFs. It is well known that the ion polarization theory exists in the ion system in the presence of EEF, and the polarization properties of water molecules with EEFs have been investigated by MD simulation method43,44, in which the polarization is enforced and the mean dipole moment of water molecules increases under a threshold electric field.Here, we think that the polarization of [Emim][PF6] IL enhances with the increase of EEFs and thus the total dipole moment of each anion and cation in the simulation system increases, which may cause the intensity of vibration mode of cation-anion to enhance as the varied EEFs increase. The total dipole moment for per IL is given asq

whereμis the total dipole moment for per anion and cation,qiis the charge amount ofith atom of anion and cation,riis the displacement forith atom, andrcom is the centroid displacement for the corresponding anion and cation. As shown in Fig. 4, we calculate the change of the total dipole moment for per anion and cation with the increase of the EEFs, and we find that the total dipole moment gradually increases from 4.34 to 5.46 Debye (D)when the EEF increases. And it is worth noting that the dipole moment obtained from a polarizable force field simulation is reliable as the EEF is small, while there may be some deviation in the result when the EEF is greater than 5 V·nm−1, but this deviation is acceptable. On the other hand, the redshift phenomenon is observed for the 50 cm−1band, which first redshift 16 cm−1when the EEF changes from 0 to 2 V·nm−1and then the shift wavenumber increases to 33 cm−1with the EEF in the range from 3 to 10 V·nm−1. Ludwig and co-workers16have observed a similar redshift trend for cation-anion vibration mode which was explained as a decrease in interaction energy between anions and cations. In our work, we calculate and analyze the average interaction energy between anions and cations in the simulation system to give a reasonable explanation for the redshift phenomenon in Fig. 5. As can be seen in this figure, with the varied EEFs increase from 0 to 10 V·nm−1, the average Coulombic interaction energy decreases from −307.6 to −269.2 kJ∙mol−1and the average vdW interaction energy reduces from−71.1 to −28.8 kJ∙mol−1. Therefore, the average interaction energy for per ion of [Emim][PF6] IL shows decrease with the increasing EEFs, and the 50 cm−1VB with respect to the cationanion vibration mode exhibits redshift feature. Under the action of EEFs, the cation as a whole move along the direction of the EEFs, while the anion moves along the opposite direction of the EEFs. So we calculate the average distance between 512 anions and cations as shown in Fig. S2 (in Supporting Information), the phenomenon can be found that the average distances gradually increase from 0.632 to 0.789 nm with the EEFs increase from 0 to 10 V·nm−1. It further illustrates that the cation and anion move with the EEFs, and the average distances between anion and cation gradually increase resulting in the average interaction energy for per IL gradually decrease.

Fig. 4 The change of the total dipole moment of each anion and cation of [Emim][PF6] IL under varied EEFs ranging from 0 to 10 V·nm−1.

Fig. 5 Average interaction energy for per ion of [Emim][PF6] IL with the varied EEFs in the range of 0 to 10 V·nm−1 for (a)average Coulombic energy (Ecoul)and vdw energy (Evdw)and (b)total binding energy (Ebin = Ecoul + Evdw)for per [Emim][PF6] ion.
For the 183 cm−1VB which is attributed to the torsional vibration mode of ethyl side chain on cationic imidazole ring,the intensity of the band slightly enhances with the EEFs increase. The previous study has confirmed that the IL structure would be reordered into an ordered structure when the applied electric field strength increases27. Herein, we think that the torsional motion of the ethyl side chain enhances with the process of the IL structure from disorder to order, and the intensity of the torsional vibration mode of ethyl side chain enhances as the EEFs varied from 0 to 10 V·nm−1. The orientation functions of the cations are calculated to prove the enhancement of torsional motion of the ethyl side chains. As shown in Fig. 6, the values of the second Legendre polynomials(P2cos(θ)= (3cos2θ− 1)/2)about the orientation angles, which is defined as the angle between the vector of nitrogen atom on cationic imidazole ring and terminal carbon atom on ethyl chain and theyaxis direction vector that is in accordance with the EEF direction, obviously increase with the varied EEFs increasing and almost are close to 0.5 as the EEF over 8 V·nm−1. This strongly suggests that the increased EEFs cause the ethyl chain of cations gradually moving toward the direction of the electric field and the average orientation of the cationic ethyl side chains gradually changing from disorder to order. So the conclusion can be summarized that the EEFs indeed enhance the twisting amplitude of the ethyl side chain, and thus enhancing the intensity of 183 cm−1band.

Fig. 6 Average values of P2cos(θ)for the orientation angles between the selected vector (starting with N atom on cationic imidazole ring and ending with thermal carbon atom on ethyl side chain)and the y axis direction vector which is in line with the EEF direction in the system with varied EEFs ranging from 0 to 10 V·nm−1, which are displayed by a three-dimensional graph and the colored shadows are the area between the curves and the baseline (P2cos(θ)= 0).
The VB at 3196 cm−1is assigned to the stretching vibration modes of the C―H bonds on the methyl (CH3―)and ethyl(CH3CH2―)side chains. As presented in Fig. 3b, the intensity of the VB is weakened with the increasing EEFs and this weakening trend is relatively obvious when the EEFs are between 1 and 5 V·nm−1. In order to visually explain the weakening of the VB strength, the radial distribution functions(RDFs)between carbon and hydrogen atoms on the methyl and ethyl side chains are calculated and shown in Fig. 7. From Fig.7 we can get almost the opposite change law, in which the peaks of three different RDFs all increase with the EEFs increase and the significant increasing trends are observed as the EEFs varying from 1 to 5 V·nm−1. So we think the RDFs between carbon atoms and hydrogen atoms increase and thus there may be more hydrogen atoms around the carbon atoms on the methyl and ethyl side chains. Thus this makes free space around the C―H bonds on the methyl and ethyl side chains reduce. The stretching vibrations of C―H bonds subject to certain restrictions with the varied EEFs increasing and the amplitude of the stretching vibrations is weakened. To further explain the increase of the number of hydrogen atoms around carbon atoms on the methyl and ethyl side chains, we calculate the coordination numbers (CNs)between carbon and hydrogen atoms on the methyl and ethyl side chains with the EEFs varying from 0 to 10 V·nm−1, presented in Fig. 8. As we can see, the CNs between carbon and hydrogen atoms on side chains all increase with the EEFs increasing, and the CNs increase significantly when the EEF varies from 1 to 5 V·nm−1. It indicates that there are indeed more hydrogen atoms around carbon atoms on the methyl and ethyl side chains with EEFs. Therefore, the intensity of the stretching vibration modes of C―H bonds on the methyl and ethyl side chains is gradually weakened as the EEFs increase.

Fig. 7 RDFs between carbon atoms and hydrogen atoms on the methyl and ethyl side chains with varied EEFs in the range from 0 to 10 V·nm−1 for (a)RDF of C―H bonds on methyl side chain,(b)RDF of C―H bonds for methylene group on ethyl side chain and(c)RDF of C―H bonds for methyl group on ethyl side chain,where the inset shows the local enlarged view.


Fig. 8 The coordination numbers (CNs)between carbon atoms and hydrogen atoms on the methyl and ethyl side chains with varied EEFs in the range from 0 to 10 V·nm−1 for (a)CN of C―H on methyl side chain, (b)CN of C―H for methylene group on ethyl side chain and (c)CN of C―H for methyl group on ethyl side chain.
As for the VB at 3396 cm−1, it is the characteristic peak of the stretching vibration mode of C2―H2, C4―H4 and C5―H5 bonds on cationic imidazole ring and the band is also as a signature of HBs in IL system45. From Fig. 3b, we can find that the intensity of the band is gradually weakened and the redshift phenomena occur at the same time. It is already known that the C―H bonds on the imidazole ring can bond with the fluorine atoms in the [PF6]−anions to form+C―H··F−HBs and the stretching vibration mode for C―H bonds straightforwardly exhibits the strength of the relevant HBs11. Thus we investigate the HBs in the following to give a better understanding for the change of the VB at 3396 cm-1. The formation of the+C―H··F−HBs is defined according to the following geometric (distance and angular)criteria:
where C is the C2, C4 and C5 atoms of the [Emim]+donor and F is the fluorine atom of the [PF6]−acceptor.RCFis the distance between the C (C2, C4 and C5 atoms)and F ([PF6]−)atoms,whileθFCHis the F··C―H angle. Then,are the distance and angle threshold values of HB formation,respectively. Based on the previously reported research14,46,47,thevalue is usually fixed at 30° andvalues are separately set as 0.38 nm (for+C2―H2··F−HB)and 0.395 nm(for+C4―H4··F−and+C5―H5··F−HBs)in this paper, which are obtained from the first minimum of the corresponding RDFs of the bulk [Emim][PF6] IL at 350 K and 0 V·nm−1EEF (Fig. S3,in Supporting Information). The change of the HB numbers with EEFs varying from 0 to 10 V·nm−1is shown in Fig. 9. We see that the whole number of HBs is reduced as the EEFs increase and the ability of C2 atoms to form HBs is stronger than C4 and C5 atoms. This is consistent with the results obtained in the literature48that the C4―H and C5―H groups involve weak HBs and the C2―H groups form stronger HBs. So the intensity of the stretching vibration mode of C―H bonds on cationic imidazole ring is weakened with the increasing EEFs. In addition, the intensity remains basically unchanged when the EEF is greater than 4 V·nm−1. Generally speaking, the corresponding VB should redshift as the HB increasing.However, an anomalous phenomenon appears in our research work and the VB is weakened and redshifted with the HB decreasing. In order to further explore the reasons for this anomaly occurs, the RDFs between hydrogen atoms (H2, H4 and H5 atoms)on cationic imidazole ring and fluorine atom in [PF6]−anions are calculated and presented in Fig. 10. As can be seen from Fig. 10, the first peaks of RDFs all shift to the left and their values become smaller as the EEFs increasing. The phenomenon may be referred to as a relaxation effect25,49,50and it would limit the ion mobility. As previously mentioned, the IL structure gradually becomes ordered with the EEFs increase. So we think the ordered structure may restrict the movement of ions, but it cannot completely prevent the effect of EEFs on cations and anions in the system. The result of this effect is that the increasing tendency for the distance between anions and cations gets slower (Fig. S2, in Supporting Information)and the first peaks of RDFs between hydrogen and fluorine atoms shift to the left. Therefore, the EEFs break the existing HB network and decrease the number of HB, meanwhile the relaxation effect results in the shortening of the distance between fluorine and hydrogen atoms and is beneficial to the HBs formation. From Fig. 9, we can get that the decrease tendency of numbers of HBs becomes slower with EEF above 3 V·nm−1and the total HBs numbers slightly increase when the EEF reaches a critical value of 8 V·nm−1. In summary, the intensity of the stretching vibration mode of C―H bonds is weakened and the peak redshifts with the EEFs increasing due to the decrease of the numbers of+C―H··F−HBs and the shortening of the distance between fluorine and hydrogen atoms caused by the relaxation effect under the varied EEFs.

Fig. 9 The numbers of +C―H··F− HBs formed by C―H bonds on cationic imidazole rings and fluorine atoms in [PF6]− anions with varied EEFs in the range of 0 to 10 V·nm−1 from (a)+C2―H2··F−HBs, +C4―H4··F− and +C5―H5··F− HBs, respectively, and(b)total HBs in the simulation system.

Fig. 10 RDFs between hydrogen atoms on the cationic imidazole rings and fluorine atoms in [PF6]− anions with varied EEFs in the range of 0 to 10 V·nm−1 from (a)H2 atoms and fluorine atoms,(b)H4 atoms and fluorine atoms and (c)H5 atoms and fluorine atoms, respectively, where the inset shows the local enlarged view and the dashed lines represent the left shift of the RDFs’ first peaks.
4 Conclusions
In this work, we have explored the effect of a series of EEFs varying from 0 to 10 V·nm−1on the vibrational spectra of imidazole IL [Emim][PF6] in a bulk system using MD simulation, and systematically analyzed the intrinsic origin of the changes for VBs at 50, 183, 3196 and 3396 cm−1,respectively. The research results indicate that the intensity of the 50 cm−1VB constantly enhances with the EEFs increasing and the peak position redshifts 16 cm−1with 2 V·nm−1EEF and 33 cm−1with EEF beyond 3 V·nm−1, and the intensity of 183 cm−1band slightly increases as the EEFs increase. Moreover, the intensities of VBs at 3196 and 3396 cm−1are obviously weakened when the varied EEFs increase from 0 to 10 V·nm−1,and meanwhile, the 3396 cm−1band redshifts 16 cm−1with 3 V·nm−1EEF and 33 cm−1with EEFs over 4 V·nm−1. Further analysis shows that the total dipole moment of each anion and cation gradually increases from 4.34 to 5.46 D and the average interaction energy for per ion of [Emim][PF6] IL decreases from−378.7 to −298.0 kJ∙mol−1when the EEF increases from 0 to 10 V·nm−1, which results in the changes of the intensity and position of VB at 50 cm−1associated with the cation-anion vibration mode. The intensity of VB at 183 cm−1assigned to the torsional vibration mode of ethyl side chain is slightly enhanced because the EEFs increase the twisting of ethyl side chain and the orientations of cations in the system are more consistent. In regard to the stretching VB for C―H bonds on methyl and ethyl side chains at 3196 cm−1, the intensity is weakened due to that more hydrogen atoms appear around the carbon atoms on the methyl/ethyl chains, and thus the stretching vibrations of C―H bonds are limited with the varied EEFs increasing. The changes of intensity and position of VB at 3396 cm−1, assigned as the C―H bonds stretching vibration mode of cationic imidazole ring, present an anomalous phenomenon, which could be caused by the decrease of average+C―H··F−HBs numbers in the system and the shorting of the distance between hydrogen and fluorine atoms that arises from the relaxation effect under EEFs to the benefit of the formation of HBs. The result of the synergy of these factors leads to the obvious weakening of band intensity and redshift of peak position. The research results in this paper could be extremely important for opening outlooks in exploring the EEF effects on physicochemical properties of new IL materials.
Supporting Information:available free of chargeviathe internet at http://www.whxb.pku.edu.cn.

- 物理化学学报的其它文章
- Hollow Nitrogen-Rich Carbon Nanoworms with High Activity for Metal-Free Selective Aerobic Oxidation of Benzyl Alcohol
- Photocrosslinking-Immobilized Polymer Vesicles for Lowering Temperature Triggered Drug Release
- CO Hydrogenation to Ethanol over Supported Rh-Based Catalyst:Effect of the Support
- CdTeSe合金幻数团簇的室温合成和形成机理研究
- 体相界面导通的复合快离子导体
- 异氰酸苯酯诱导的类胶原多肽自组装