
抗结核新药研发进展
2021-11-18 08:55吴姗姗
上海医药 2021年21期
吴姗姗
摘 要 结核病由结核分枝杆菌感染引起,是一类慢性、传染性和致命性的疾病。随着耐多药和广泛耐药结核分枝杆菌的不断出现与快速传播,结核病治疗变得更加艰难,寻找新药成为当务之急。本文介绍近年来抗结核新药的研发进展。
关键词 抗结核药物 结核病 结核分枝杆菌
中图分类号:R978.3 文献标志码:A 文章编号:1006-1533(2021)21-0011-05
Research progress of new antituberculosis drugs
WU Shanshan
(Department of Pharmacy, Shanghai Public Health Clinical Center, Shanghai 201508, China)
ABSTRACT Tuberculosis is caused by the Mycobacterium tuberculosis and is a chronic, infectious and fatal disease. Its treatment becomes more difficult due to the emergence and rapid spread of multidrug-resistant and extensively drug-resistant Mycobacterium tuberculosis. So it is urgent to find new drugs. The new drugs found in recent years are briefly described so as to provide a reference for the research and development of new antituberculosis drugs.
KEy wORDS antituberculosis drugs; tuberculosis; Mycobacterium tuberculosis
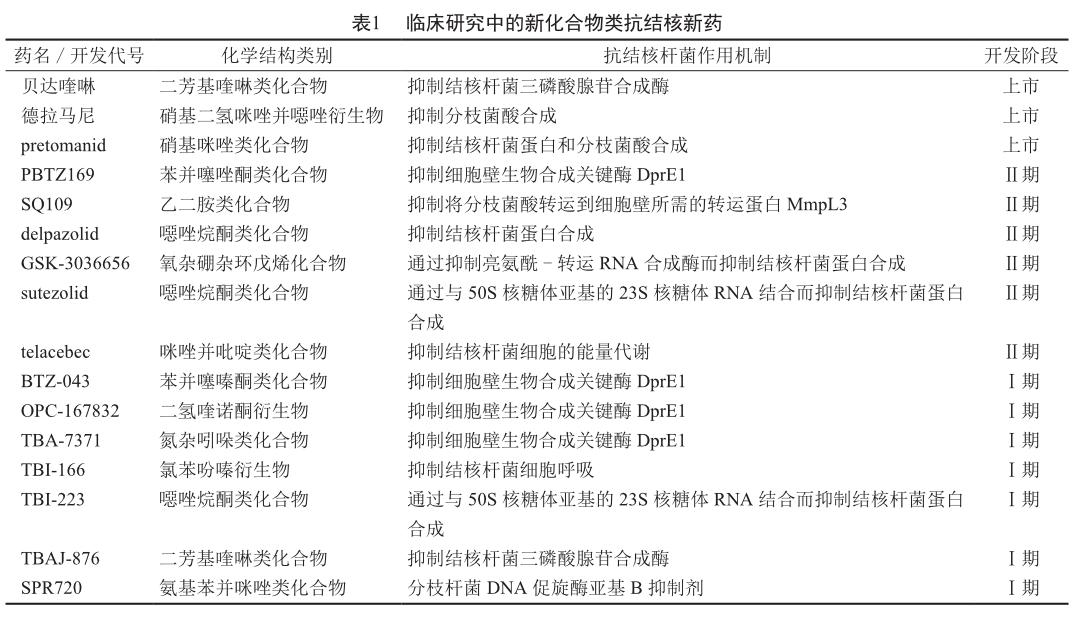
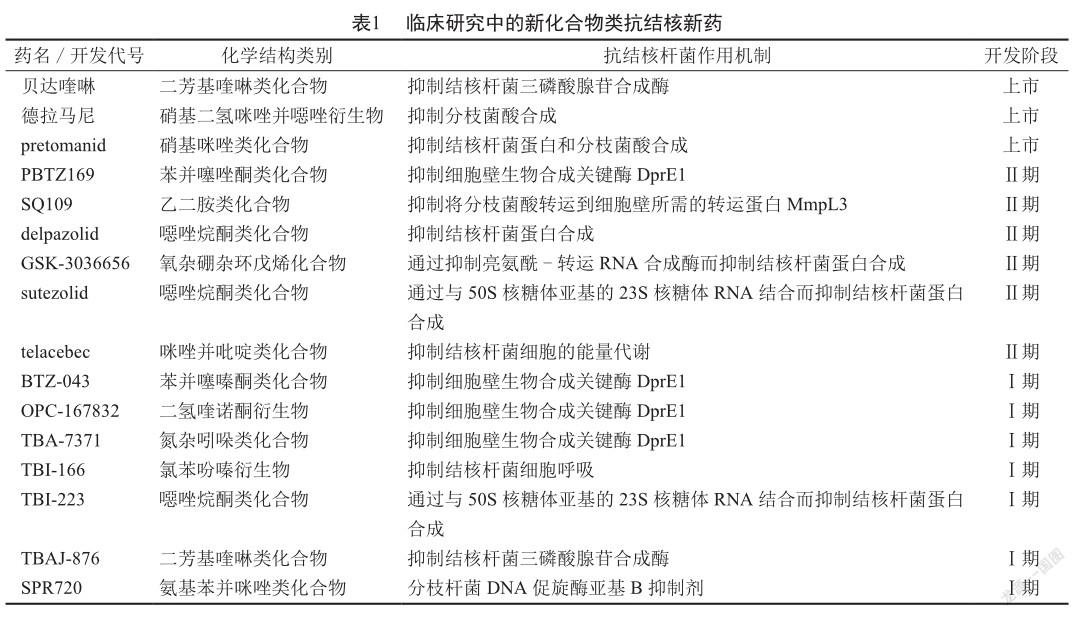
肺結核是一种传染性疾病,通常情况下仅影响肺部,但约25%患者的结核分枝杆菌(以下简称“结核杆菌”)会通过血液进入并感染身体的其他部位,如胸膜、脑膜、淋巴系统、泌尿生殖系统、骨骼和关节等。结核病可治愈、可预防。有数据显示,2019年全球结核病患者中有78%的患者为耐多药患者;61%的结核病患者被检测出对利福平耐药,而此比率在2017年为51%,在2012年为7%;约9.5%的耐多药结核病患者实为广泛耐药的患者[1-2]。因此,临床上迫切需要有对耐药结核杆菌感染有效的新药。事实上,人们也确在积极研究与开发新的抗结核药物:截至2020年8月,除已获准上市的贝达喹啉(bedaquiline)、德拉马尼(delamanid)和pretomanid外,还有19种抗结核新药处于不同临床研究阶段[3],其中包括13种新化合物和6种意欲增加抗结核新用途的已上市药物。本文就新化合物类抗结核新药(表1)的研发进展作一简要介绍。
1 已上市的抗结核新药
1.1 贝达喹啉
贝达喹啉是美国FDA自20世纪70年代末以来批准上市的第一种抗结核药物,其具有独特的作用机制,为结核杆菌三磷酸腺苷合成酶抑制剂。研究显示,使用贝达喹啉治疗的耐多药结核病患者的痰菌培养转阴率高达65% ~ 100%;贝达喹啉与利奈唑胺联用可能会提高贝达喹啉的疗效,与德拉马尼联用可将治疗2个月后的痰菌培养转阴率从29.6%提高到40%以上[4]。在耐多药结核病治疗中,加用贝达喹啉能显著改善治疗效果[5]。
贝达喹啉是抗结核联合疗法的一个组分,其最终消除半衰期明显长于其他抗结核药物,达5 ~ 6个月,加之具有较强的肝毒性,故临床上缩短治疗时间和保障用药安全非常重要。需指出的是,贝达喹啉与口服降糖药具有相同的肝脏代谢途径,所以它们同服可能发生相互作用。其中,贝达喹啉与二甲双胍的相互作用较弱,但同服时的严重胃肠道反应发生率仍可能增高 [6]。
1.2 德拉马尼
德拉马尼是硝基二氢咪唑并噁唑衍生物,其能抑制分枝菌酸的合成,体内外实验均显示对耐药结核杆菌株有很强的抗菌活性[7]。德拉马尼与抗反转录病毒药物的相互作用潜力较低,已被WHO建议用于成人耐多药结核病的治疗[8]。尽管德拉马尼治疗的总体耐受性良好,但其可导致患者QT间期延长,特别是在与其他有这种作用的抗结核药物(如氟喹诺酮类药物)联用时[9]。德拉马尼对QT间期的影响多发生在开始用药后的最初6 ~ 10周内,且与其用药剂量相关。这与德拉马尼的主要代谢物DM-6705有关。细胞色素P450酶(cytochrome P450, CYP)3A4诱导剂和抑制剂都可能提高DM-6705的水平。德拉马尼与贝达喹啉和/或其他具有QT间期延长作用的抗结核药物(如氟喹诺酮类药物、氯法齐明)联用时容易导致患者发生潜在有害的QT间期延长,故WHO建议,在开始这种联合疗法前应评估患者的心电图,并在治疗期间对患者的心电图进行定期监测[8]。
德拉马尼已在临床上广泛用于成人耐多药结核病患者的治疗,用于儿童耐多药结核病患者的研究也越来越多。研究显示,使用德拉马尼治疗2个月可降低儿童广泛耐药结核病患者的痰菌培养阳性率[10]。德拉马尼治疗儿童耐药结核病患者有效、安全、耐受性良好,唯一令人不安的不良反应是QT间期延长,发生率约为10%,具有明显的剂量相关性。
1.3 pretomanid
pretomanid为硝基咪唑类化合物。体内外实验显示,pretomanid对结核杆菌有较强的杀菌活性,抑制90%结核杆菌生长的最低抑制质量浓度(minimum inhibitory concentration, MIC)(MIC90)为0.12 μg/mL[11]。pretomanid是美国FDA近40年来批准的第3种抗结核药物,其被批准与贝达喹啉和利奈唑胺联用,治疗特定高度耐药的肺结核患者。在高度耐药结核病患者中,90%的患者在使用pretomanid、贝达喹啉和利奈唑胺治疗6个月后获得良好的疗效,同时没有数据表明结核杆菌对pretomanid出现了耐药[12]。
2 研发中的抗结核新药
2.1 BTZ-043
DprE1是结核杆菌细胞壁生物合成的关键酶,与DprE2共同催化转化D-阿拉伯呋喃糖为阿拉伯聚糖,而阿拉伯聚糖是结核杆菌细胞壁的必要组分。因此,DprE1已被用作一种新的抗结核药物作用靶点。BTZ-043是一种苯并噻嗪酮类化合物,能通过抑制DprE1而抑制结核杆菌细胞壁的合成[13]。结核病动物模型研究显示,BTZ-043对结核杆菌的MIC为1 ng/mL,较现使用的任何抗结核药物都更有效[14-15]。此外,迄今为止所有的结核杆菌临床分离株,包括耐多药和广泛耐药结核杆菌株,均对BTZ-043敏感,表明BTZ-043是一个有发展前途的候选抗结核药物[16]。
2.2 PBTZ169
PBTZ169为苯并噻唑酮类化合物,其能通过抑制DprE1而抑制結核杆菌细胞壁的合成,对对药物敏感和耐药结核杆菌株的MIC均为1 μmol/L,与其他抗结核药物有协同作用,具有潜在的杀菌作用,可显著缩短结核病复发小鼠模型的治疗时间[17]。PBTZ169的细胞毒性较BTZ-043小,较低血药浓度下即对结核病小鼠模型有很好的疗效,这可能与PBTZ169能较BTZ-043更有效地抑制DprE1有关。与BTZ-043治疗组相比,PBTZ169治疗组小鼠的肺部细菌负荷显著减少。PBTZ169治疗结核病与异烟肼一样有效,值得进一步研究[18]。
2.3 OPC-167832
OPC-167832为二氢喹诺酮衍生物,其作用靶点也是结核杆菌细胞壁生物合成的关键酶DprE1,对细胞内结核杆菌H37Rv株和Kurono株的MIC90分别为0.004 8和0.002 7 μg/mL,低于利福平的1.150 1和0.564 8 μg/mL[19]。OPC-167832在低质量浓度下即对细胞内结核杆菌株有杀菌作用,对结核杆菌的MIC为0.000 24 ~ 0.002 μg/mL,对生长菌和胞内菌均有杀菌活性[19]。这些研究结果提示,OPC-167832是一种有发展前途的候选抗结核药物。
2.4 TBA-7371
TBA-7371为氮杂吲哚类化合物,其能通过抑制DprE1而抑制结核杆菌细胞壁的合成,对结核杆菌的MIC为0.28 ~ 1.12 μg/mL,并已经结核病啮齿动物模型研究显示具有抗结核病作用[20]。
2.5 SQ109
SQ109为乙二胺类化合物,其能抑制结核杆菌的膜蛋白MmpL3,而MmpL3是将分枝菌酸转运到细胞壁所需的转运蛋白[21]。SQ109具有良好的抗结核杆菌活性[22],对细胞内结核杆菌的抑制作用较sutezolid强[23]。
2.6 delpazolid(LCB01-0371)
delpazolid为噁唑烷酮类化合物,其能通过与细菌50S核糖体亚基的23S核糖体RNA的V结构域结合来抑制信使RNA的转录,从而抑制结核杆菌等细菌蛋白的合成[24]。
2.7 GSK-3036656
亮氨酰-转运RNA合成酶(leucyl-tRNA synthetase, LeuRS)是结核杆菌蛋白合成过程中的关键酶,许多LeuRS抑制剂都具有良好的抗结核杆菌活性。GSK- 3036656是一种新型氧杂硼杂环戊烯化合物,其能高度选择性地作用于结核杆菌LeuRS,在结核病小鼠模型中表现出显著的抗结核作用[25]。研究显示,GSK-3036656对结核杆菌LeuRS有很强的抑制作用(MIC50为0.20μmol/L)和体外抗结核杆菌活性(对结核杆菌H37Rv株的MIC为0.08 μmol/L)。GSK-3036656治疗的总体耐受性良好,未出现严重不良事件[25]。
2.8 sutezolid(PNU-100480)
噁唑烷酮类化合物是RNA抑制剂,它们能通过与50S核糖体亚基的23S核糖体RNA结合而抑制结核杆菌蛋白的合成。sutezolid和利奈唑胺均为噁唑烷酮类化合物,它们都能通过抑制细菌蛋白合成中的70S起始复合物的合成而抑制信使RNA的转录,均有抗结核杆菌活性,包括抗耐多药和广泛耐药菌株活性[26-27]。sutezolid与利奈唑胺对结核杆菌的MIC相近,但sutezolid的抗结核作用优于利奈唑胺,无论是单用还是与其他抗结核药物联用[26-27]。
2.9 TBI-166
TBI-166的化学结构与氯法齐明相似,但其抗结核杆菌活性优于氯法齐明[28],对结核杆菌的MIC为0.016μg/mL[29]。一项研究在小鼠气溶胶感染模型中比较了5种抗结核治疗方案的杀菌活性,结果显示TBI-166与贝达喹啉和利奈唑胺联用方案的杀菌活性最强[30]。
2.10 TBI-223
TBI-223为噁唑烷酮类化合物,其能抑制结核杆菌蛋白的合成[31]。
2.11 TBAJ-876
TBAJ-876除具有抗结核杆菌活性外,还具有抗脓肿分枝杆菌活性,这意味着其不仅可能对肺结核治疗有用,且也可能对由脓肿分枝杆菌感染引起的疾病治疗有用[32]。贝达喹啉存在与QT间期延长相关的心脏毒性。因此,尽管贝达喹啉对耐药结核病有很好的治疗效果,但其应用仍因潜在严重不良反应而受到一定的限制。为了解决这一问题,人们找到了贝达喹啉的改良类似物TBAJ-876[33]。结核病动物模型研究显示,与贝达喹啉相比,TBAJ-876具有更强的抗结核杆菌活性,故有降低临床用药剂量以提高安全性的潜力[34]。
2.12 SPR720
SPR720是SPR719的口服前藥,用于治疗肺部非结核杆菌分枝杆菌感染[35]。体外实验显示,SPR719对多种非结核杆菌分枝杆菌有抗菌活性,其对几种非结核杆菌分枝杆菌株的MIC50为0.25 ~ 4 μg/mL[36]。
2.13 telacebec(Q203)
telacebec为咪唑并吡啶类化合物,其能通过抑制结核杆菌细胞色素bc1复合物来干扰结核杆菌细胞能量的产生,体内外实验均发现对结核杆菌有抑制作用[37]。telacebec对结核杆菌H37Rv株的MIC为2.7 nmol/L,对耐多药和广泛耐药结核杆菌株的MIC分别为0.28和0.43 nmol/L,抗结核杆菌活性较强[38]。
3 结语
肺结核治疗多采用多种药物联合用药,且用药持续时间长,其中耐多药结核病患者常需持续用药6 ~ 20个月。结核病治疗面临的挑战首先是治疗方案的复杂性和用药持续时间长,两者都会影响患者的治疗依从性;其次是不良反应,尤其是用于治疗耐药结核病的二线治疗药物的不良反应较多,且抗结核药物与其他药物之间的相互作用情况也较复杂。因此,临床上迫切需要有更有效、更安全的抗结核新药。抗结核新药的研发进展令人关注。
参考文献
[1] Dalberto PF, de Souza EV, Abbadi BL, et al. Handling the hurdles on the way to anti-tuberculosis drug development [J]. Front Chem, 2020, 8: 586294.
[2] Oh S, Trifonov L, Yadav VD, et al. Tuberculosis drug discovery: a decade of hit assessment for defined targets [J]. Front Cell Infect Microbiol, 2021, 11: 611304.
[3] WHO. Global tuberculosis report 2020 [EB/OL]. [2021-03-23]. https://apps.who.int/iris/bitstream/handle/10665/ 336069/9789240013131-eng.pdf?sequence=1&isAllowed=y.
[4] Li Y, Sun F, Zhang W. Bedaquiline and delamanid in the treatment of multidrug-resistant tuberculosis: promising but challenging [J]. Drug Dev Res, 2019, 80(1): 98-105.
[5] Degiacomi G, Sammartino JC, Sinigiani V, et al. In vitro study of bedaquiline resistance in Mycobacterium tuberculosis multi-drug resistant clinical isolates [J]. Front Microbiol, 2020, 11: 559469.
[6] Hu M, Zheng C, Gao F. Use of bedaquiline and delamanid in diabetes patients: clinical and pharmacological considerations[J]. Drug Des Devel Ther, 2016, 10: 3983-3994.
[7] Nieto Ramirez LM, Quintero Vargas K, Diaz G. Whole genome sequencing for the analysis of drug resistant strains of Mycobacterium tuberculosis: a systematic review for bedaquiline and delamanid [J]. Antibiotics (Basel), 2020, 9(3): 133.
[8] WHO. The use of delamanid in the treatment of multidrugresistant tuberculosis: interim policy guidance [EB/ OL]. [2021-03-23]. http://apps.who.int/iris/bitstream/ handle/10665/137334/WHO_HTM_TB_2014.23_eng. pdf?sequence=1.
[9] Szumowski JD, Lynch JB. Profile of delamanid for the treatment of multidrug-resistant tuberculosis [J]. Drug Des Devel Ther, 2015, 9: 677-682.
[10] Esposito S, Bosis S, Tadolini M, et al. Efficacy, safety, and tolerability of a 24-month treatment regimen including delamanid in a child with extensively drug-resistant tuberculosis: a case report and review of the literature [J]. Medicine (Baltimore), 2016, 95(46): e5347.
[11] Zhang F, Li S, Wen S, et al. Comparison of in vitro susceptibility of mycobacteria against PA-824 to identify key residues of Ddn, the deazoflavin-dependent nitroreductase from Mycobacterium tuberculosis [J]. Infect Drug Resist, 2020, 13: 815-822.
[12] Gils T, Lynen L, de Jong BC. Pretomanid for tuberculosis: a systematic review [J]. Clin Microbiol Infect, 2021, 13: S1198-743X(21)00464-X.
[13] Lienhardt C, Raviglione M, Spigelman M, et al. New drugs for the treatment of tuberculosis: needs, challenges, promise, and prospects for the future [J]. J Infect Dis, 2012, 205(Suppl 2): S241-S249.
[14] Neres J, Pojer F, Molteni E, et al. Structural basis for benzothiazinone-mediated killing of Mycobacterium tuberculosis [J]. Sci Transl Med, 2012, 4(150): 150ra121.
[15] Makarov V, Manina G, Mikusova K, et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis [J]. Science, 2009, 324(5928): 801-804.
[16] Wolucka BA. Biosynthesis of D-arabinose in mycobacteria—a novel bacterial pathway with implications for antimycobacterial therapy [J]. FEBS J, 2008, 275(11): 2691-2711.
[17] Zhang G, Howe M, Aldrich CC. Spirocyclic and bicyclic 8-nitrobenzothiazinones for tuberculosis with improved physicochemical and pharmacokinetic properties [J]. ACS Med Chem Lett, 2019, 10(3): 348-351.
[18] Makarov V, Lechartier B, Zhang M, et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones [J]. EMBO Mol Med, 2014, 6(3): 372-383.
[19] Hariguchi N, Chen X, Hayashi Y, et al. OPC-167832, a novel carbostyril derivative with potent antituberculosis activity as a DprE1 inhibitor [J]. Antimicrob Agents Chemother, 2020, 64(6): e02020-19.
[20] Working Group on New TB Drugs. TBA-7371 [EB/ OL]. [2021-03-23]. https://www.newtbdrugs.org/pipeline/ compound/tba-7371.
[21] Tahlan K, Wilson R, Kastrinsky DB, et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis [J]. Antimicrob Agents Chemother, 2012, 56(4): 1797-1809.
[22] Nikonenko BV, Protopopova M, Samala R, et al. Drug therapy of experimental tuberculosis (TB): improved outcome by combining SQ109, a new diamine antibiotic, with existing TB drugs [J]. Antimicrob Agents Chemother, 2007, 51(4): 1563-1565.
[23] Reddy VM, Dubuisson T, Einck L, et al. SQ109 and PNU-100480 interact to kill Mycobacterium tuberculosis in vitro[J]. J Antimicrob Chemother, 2012, 67(5): 1163-1166.
[24] Choi Y, Lee SW, Kim A, et al. Safety, tolerability and pharmacokinetics of 21 day multiple oral administration of a new oxazolidinone antibiotic, LCB01-0371, in healthy male subjects [J]. J Antimicrob Chemother, 2018, 73(1): 183-190.
[25] Tenero D, Derimanov G, Carlton A, et al. First-time-inhuman study and prediction of early bactericidal activity for GSK3036656, a potent leucyl-tRNA synthetase inhibitor for tuberculosis treatment [J]. Antimicrob Agents Chemother, 2019, 63(8): e00240-19.
[26] Williams KN, Stover CK, Zhu T, et al. Promising antituberculosis activity of the oxazolidinone PNU-100480 relative to that of linezolid in a murine model [J]. Antimicrob Agents Chemother, 2009, 53(4): 1314-1319.
[27] Wallis RS, Jakubiec W, Kumar V, et al. Biomarker-assisted dose selection for safety and efficacy in early development of PNU-100480 for tuberculosis [J]. Antimicrob Agents Chemother, 2011, 55(2): 567-574.
[28] Zhu H, Fu L, Wang B, et al. Activity of clofazimine and TBI-166 against Mycobacterium tuberculosis in different administration intervals in mouse tuberculosis models [J]. Antimicrob Agents Chemother, 2021, 65(4): e02164-20.
[29] Zhang D, Lu Y, Liu K, et al. Identification of 1ess lipophilic riminophenazine derivatives for the treatment of drugresistant tuberculosis [J]. J Med Chem, 2012, 55(19): 8409-8417.
[30] Zhang Y, Zhu H, Fu L, et al. Identifying regimens containing TBI-166, a new drug candidate against Mycobacterium tuberculosis in vitro and in vivo [J]. Antimicrob Agents Chemother, 2019, 63(7): e02496-18.
[31] Working Group on New TB Drugs. TBI-223 [EB/OL]. [2021-03-23]. https://www.newtbdrugs.org/pipeline/compound/tbi-223.
[32] Sarathy JP, Ganapathy US, Zimmerman MD, et al. TBAJ-876, a 3,5-dialkoxypyridine analogue of bedaquiline, is active against Mycobacterium abscessus [J]. Antimicrob Agents Chemother, 2020, 64(4): e02404-19.
[33] Choi PJ, Conole D, Sutherland HS, et al. Synthetic studies to help elucidate the metabolism of the preclinical candidate TBAJ-876—a less toxic and more potent analogue of bedaquiline [J]. Molecules, 2020, 25(6): 1423.
[34] Sarathy JP, Ragunathan P, Cooper CB, et al. TBAJ-876 displays bedaquiline-like mycobactericidal potency without retaining the parental drugs uncoupler activity [J]. Antimicrob Agents Chemother, 2020, 64(2): e01540-19.
[35] Stokes SS, Vemula R, Pucci MJ. Advancement of GyrB inhibitors for treatment of infections caused by Mycobacterium tuberculosis and non-tuberculous mycobacteria [J]. ACS Infect Dis, 2020, 6(6): 1323-1331.
[36] Brown-Elliott BA, Rubio A, Wallace RJ Jr. In vitro susceptibility testing of a novel benzimidazole, SPR719, against nontuberculous mycobacteria [J]. Antimicrob Agents Chemother, 2018, 62(11): e01503-18.
[37] de Jager VR, Dawson R, van Niekerk C, et al. Telacebec(Q203), a new antituberculosis agent [J]. N Engl J Med, 2020, 382(13): 1280-1281.
[38] Pethe K, Bifani P, Jang J, et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis [J]. Nat Med, 2013, 19(9): 1157-1160.
猜你喜欢
保健医苑(2022年5期)2022-06-10
医学概论(2022年4期)2022-04-24
国际呼吸杂志(2019年1期)2019-03-08
中国实用医药(2016年18期)2016-08-03
中国实用医药(2016年9期)2016-05-17
中国卫生(2015年1期)2015-11-16
中外医疗(2015年3期)2015-08-29
西南军医(2015年3期)2015-04-23
中国民族民间医药·下半月(2014年6期)2015-01-21
结核与肺部疾病杂志(2014年1期)2014-07-18
