
高效液相色谱法同时测定鱼腥草芩蓝合剂中7 种成分
2021-11-13 08:36昝珂周颖郑成
化学分析计量 2021年10期
昝珂,周颖,郑成
(1.中国食品药品检定研究院,北京 102629;2.浙江省食品药品检验研究院,国家药品监督管理局中成药质量评价重点实验室,杭州 310052)
鱼腥草芩蓝合剂为常见中药制剂,由鱼腥草、黄芩、板蓝根、连翘、金银花5 味中药材配制而成,具有清热解毒的作用,临床常用于治疗外感风热引起的咽喉肿痛,急性咽炎、扁桃体炎等疾病。现代药理研究表明,该制剂具有良好的解热作用,临床治疗小儿疱疹性咽峡炎有较好的疗效[1-4]。鱼腥草芩蓝合剂收录于国家药品监督管理局国家中成药标准中,然而现行标准中只有鱼腥草、黄芩、连翘和金银花的薄层色谱鉴别项和黄芩苷含量的测定项[5],不能全面地反映复方制剂的内在质量。目前有关该制剂组分含量的测定研究较少,邓茂芳等[6]建立了高效液相色谱(HPLC)法同时测定鱼腥草芩蓝合剂中绿原酸、黄芩苷和黄芩素含量的方法。鱼腥草芩蓝口服液是同方配制的兽药制剂[7],陆安等[8]以鱼腥草芩蓝口服液为研究对象,建立了HPLC 指纹图谱鉴别方法,但未进行含量测定研究;邢玉娟等[9]建立了HPLC 法测定鱼腥草芩蓝口服液中黄芩苷和绿原酸含量的方法。鱼腥草中含有绿原酸、槲皮素、槲皮苷等化学成分[10-11];黄芩中含有黄芩苷、汉黄芩素和汉黄芩苷等成分[12-13];连翘中含有绿原酸、槲皮素、槲皮苷、连翘酯苷A 等成分[14-15];金银花中含有绿原酸、槲皮素和槲皮苷等成分[16-17]。笔者在前人研究的基础上,对试验条件进行优化,建立了HPLC 法同时测定鱼腥草芩蓝合剂中绿原酸、连翘酯苷A、黄芩苷、槲皮苷、汉黄芩苷、槲皮素和汉黄芩素7 种成分含量的方法,为全面控制该制剂的质量提供参考。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:Ultimate U3000 型,配有Chromeleon 7.2SR5 色谱工作站、真空脱气机、自动进样器、柱温箱和二极管阵列检测器,赛默飞世尔科技(中国)有限公司。
电子天平:XPE205 型,感量为0.01 mg,瑞士梅特勒-托利多公司。
超声波提取器:ELMA SONIC P 型,德国艾尔玛公司。
纯水仪:Milli PAK advantage A10 型,美国密理博公司。
复方鱼腥草合剂样品:某生产企业样品。
绿原酸、连翘酯苷A、黄芩苷、槲皮苷、汉黄芩苷、槲皮素、汉黄芩素对照品:批号分别为18071907、A19011704、18041002、19091104、18032111、19082203、18030509,纯度(质量分数)分别 为96.1%、97.2%、93.3%、100.0%、98.5%、98.8%、99.1%,成都普菲德生物技术有限公司。
乙腈、甲醇:均为色谱纯,美国赛默飞世尔公司。
磷酸,分析纯,国药集团化学试剂(北京)有限公司。
实验用水为超纯水。
1.2 溶剂配制
对照品单标储备液:分别精密称取绿原酸、连翘酯苷A、黄芩苷、槲皮苷、汉黄芩苷、槲皮素和汉黄芩素对照品各约10 mg,其中黄芩苷置于10 mL 容量瓶中,其它6 种化合物分别置于100 mL 容量瓶中,各加入50%甲醇溶液溶解,稀释,定容至标线,配制成绿原酸、连翘酯苷A、黄芩苷、槲皮苷、汉黄芩苷、槲皮素和汉黄芩素的质量浓度分别为98.5、102.4、994、97.8、101.2、98.8、103.2 μg/mL 的 单 标储备液。
混合对照品溶液:分别精密量取上述对照品单标储备液各1 mL,置于同一只10 mL 容量瓶中,加入50%甲醇溶液稀释并定容至标线,摇匀,配制成绿原酸、连翘酯苷A、黄芩苷、槲皮苷、汉黄芩苷、槲皮素、汉黄芩素的质量浓度分别为9.85、10.24、99.4、9.78、10.12、9.88、10.32 μg/mL 的混合对照品溶液。
1.3 样品处理
精密量取鱼腥草芩蓝合剂2 mL,置于20 mL棕色容量瓶中,加入50%甲醇溶液12 mL,超声20 min,冷却至室温,用50%甲醇溶液稀释至标线,摇匀,用0.45 μm 微孔滤膜过滤,取续滤液,待测。
1.4 色谱条件
色 谱 柱:Symmetry Shield RP18 柱(4.6 mm×250 mm,5 μm,美国沃特世公司);流动相:乙腈-0.1%甲酸溶液;洗脱方式:梯度洗脱;洗脱程序:初始乙腈体积分数为20%,0~30 min,乙腈由20%逐渐增加至40%,保持30 min,60~80 min,乙腈由40%逐渐增加至80%;流量:1.0 mL/min;检测波长:210 nm;柱温:30 ℃;进样体积:5 μL。
2 结果与讨论
2.1 检测波长选择
所测7 种化合物中,绿原酸为咖啡酰奎宁酸类,紫外光谱最大吸收峰为327 nm;连翘酯苷A 为苯乙醇苷类,最大吸收为330 nm;其余成分为黄酮或黄酮苷类,黄芩苷最大吸收波长为278 nm,槲皮苷为254 nm 和350 nm,汉黄芩苷为276 nm,槲皮素为370 nm,汉黄芩素为276 nm。各成分之间紫外最大吸收波长差别较大,但在210 nm 处均有较强的紫外吸收,故选择210 nm 作为检测波长同时检测7种成分。
2.2 色谱条件优化
分别选择乙腈-水溶液、乙腈-0.1%甲酸溶液和乙腈-0.1%磷酸溶液作为流动相,考察不同流动相体系对7 种待测成分的分离效果。结果表明,乙腈-0.1%磷酸溶液作为流动相时,色谱峰形较好,故选择乙腈-0.1%磷酸溶液为流动相。由于7 种待测化合物极性跨度较大,采用等度洗脱无法对所有化合物进行分离,故采用梯度洗脱方式。对梯度洗脱程序进行优化,最终确定洗脱程序:初始乙腈体积分数为20%,0~30 min,乙腈由20%逐渐增加至40%,保持30 min,60~80 min,乙腈由40%逐渐增加至80%。在此洗脱程序下,7 种化合物在80 min 内获得较好的分离,峰形较好。
2.3 色谱柱选择
分别对SymmetryShield RP18 柱(4.6 mm×250 mm,5 μm)、Agilent ZORBAX Eclipse XDB 柱(250×4.6 mm,5 μm)和ODS HHPERSIL 柱(4.6 mm×250 mm,5 μm) 3 种型号的色谱柱进行考察,结果发现,SymmetryShield RP18(4.6 mm×250 mm,5 μm)色谱柱分离效果最佳,故选择该色谱柱进行方法学考察和样品测定。
2.4 样品提取条件选择
样品为口服液体制剂,溶解性较好,但个别化合物浓度较高,故采用50%甲醇对样品制剂稀释10 倍,过滤后直接进样测定。混合对照品溶液和样品溶液色谱图如图1 所示。
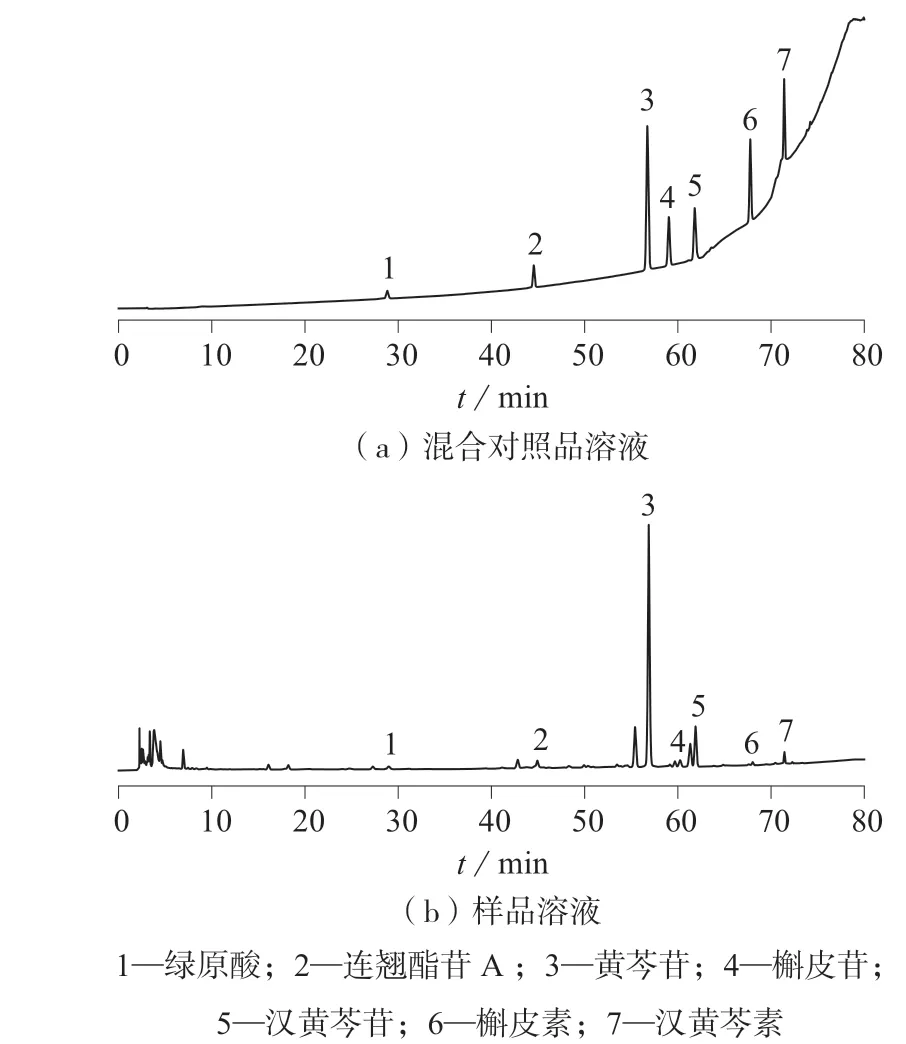
图1 混合对照品溶液和样品溶液色谱图
2.5 线性方程与检出限
精密吸取1.2 中的混合对照品溶液1、2、5、10、15、20 μL,按1.4 色谱条件分别进样测定,以待测化合物的质量浓度(x)为横坐标,色谱峰面积(y)为纵坐标进行线性回归,计算线性方程和相关系数。
将1.2 中的混合对照品溶液用50%甲醇溶液逐级稀释,在1.4 色谱条件下测定,以3 倍信噪比对应的质量浓度作为检出限,以10 倍信噪比对应的质量浓度作为定量限。7 种待测成分的线性范围、线性方程、相关系数、检出限及定量限见表1。
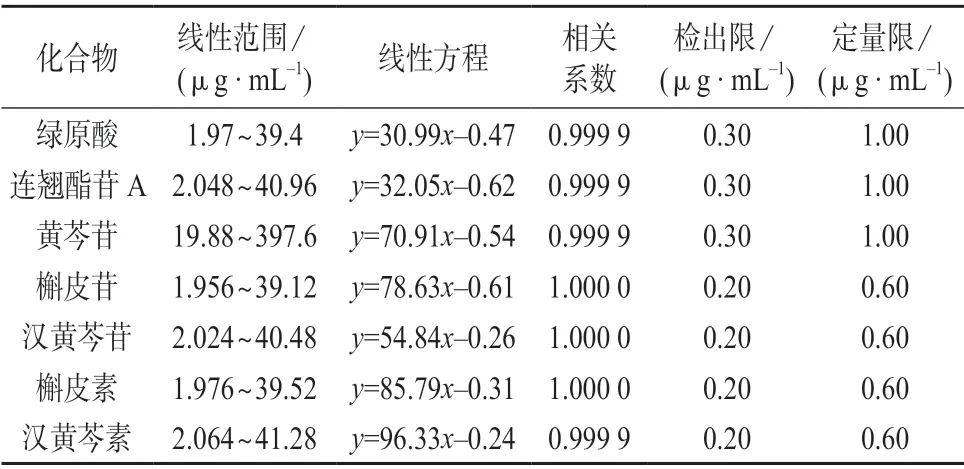
表1 7 种化合物的线性范围、线性方程、相关系数、检出限及定量限
由表1 可知,7 种待测化合物在各自的质量浓度范围内与色谱峰面积线性关系良好,相关系数均大于0.999,方法的检出限和定量限均满足鱼腥草芩蓝合剂中7 种化合物的检测需要。
2.6 稳定性试验
取鱼腥草芩蓝合剂样品,按照1.3 样品处理方法制备样品溶液,分别在配制完成后第1、2、4、8、12、24 h 进样,测定各待测化合物的色谱峰面积。绿原酸、连翘酯苷A、黄芩苷、槲皮苷、汉黄芩苷、槲皮素和汉黄芩素色谱峰面积测定结果的相对标准偏差分别为1.78%、0.81 %、0.74%、0.93%、1.21 %、1.01%、1.22%,表明样品溶液在24 h 内稳定性良好。
2.7 精密度试验
取鱼腥草芩蓝合剂样品,按1.3 方法平行制备6份样品溶液,在1.4 色谱条件下分别进样测定,计算绿原酸、连翘酯苷A、黄芩苷、槲皮苷、汉黄芩苷、槲皮素和汉黄芩素的质量浓度,结果见表2。
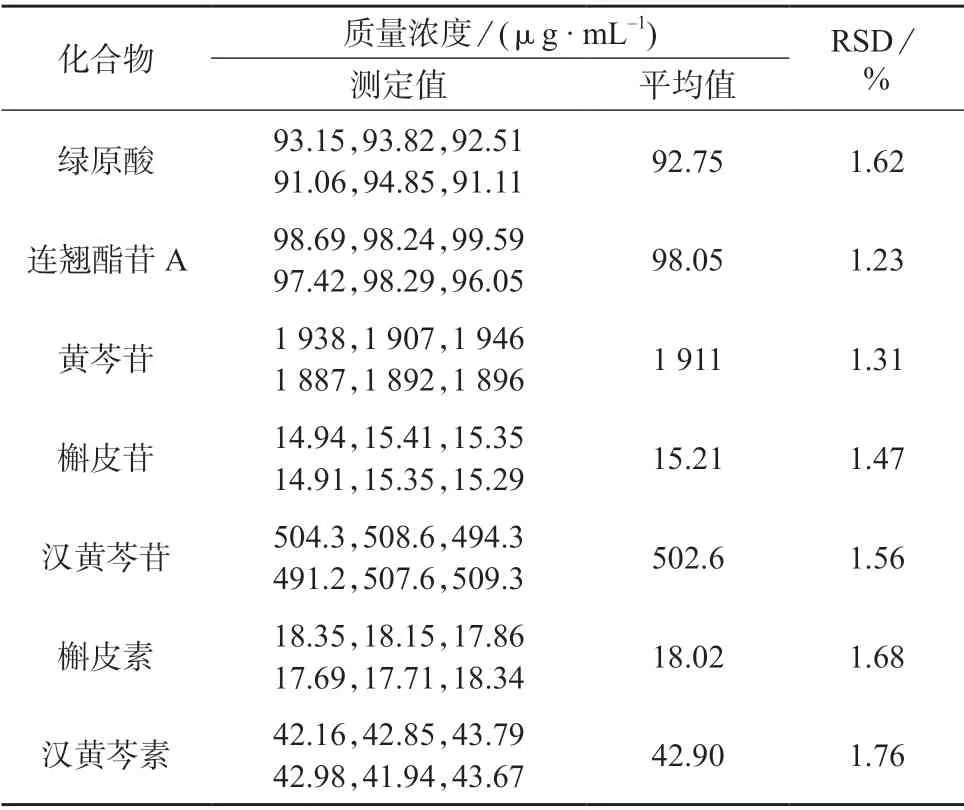
表2 精密度试验结果
由表2 可知,绿原酸、连翘酯苷A、黄芩苷、槲皮苷、汉黄芩苷、槲皮素和汉黄芩素测定结果的相对标准偏差为1.23%~1.76%,表明该方法精密度较好,满足测定要求。
2.8 加标回收试验
精密量取6 份鱼腥草芩蓝合剂样品各1 mL,分别精密加入绿原酸、连翘酯苷A、黄芩苷、槲皮苷、汉黄芩苷、槲皮素和汉黄芩素单标储备液各1.0、1.0、2.0、0.2、5.0、0.2、0.5 mL,按照1.3 样品处理方法制备加标样品溶液,在1.4 色谱条件下分别进样测定,计算样品加样回收率,结果见表3。

表3 加标回收试验结果
由表3 可知,样品加标平均回收率为在98.17%~101.48%,表明该方法准确性良好。
3 结语
建立了高效液相色谱法同时测定鱼腥草芩蓝合剂中绿原酸、连翘酯苷A、黄芩苷、槲皮苷、汉黄芩苷、槲皮素和汉黄芩素含量的方法。所测7 种成分涉及4 种药材中的活性成分,涵盖了多类指标成分,包括2 种苯丙素和5 种黄酮,为鱼腥草芩蓝合剂的质量控制提供了参考方法。
猜你喜欢
今日农业(2022年16期)2022-09-22
今日农业(2021年21期)2022-01-12
今日农业(2020年19期)2020-12-14
今日农业(2020年16期)2020-09-25
北方药学(2020年12期)2020-04-30
中华肺部疾病杂志(电子版)(2020年1期)2020-01-07
中国药理学与毒理学杂志(2019年4期)2019-08-12
农产品加工(2019年13期)2019-01-05
恋爱婚姻家庭·养生版(2018年5期)2018-05-14
中国(俄文)(2016年7期)2016-09-18
