
超高效液相色谱-串联质谱法快速测定雷公藤中毒样品中4 种毒性成分
2021-11-13 08:36:36华正罡陈曦冯静
化学分析计量 2021年10期
华正罡,陈曦,冯静
(辽宁省疾病预防控制中心,辽宁省空气雾霾与人群健康监测重点实验室,沈阳 110005)
雷公藤,别名黄藤根、断肠草、旱禾花(《湖南药物志》)、黄藤木(《广西药植名录》)、黄藤草(《江西草药手册》)[1]等,是传统的中药材。近年来,对雷公藤的基础研究主要集中在雷公藤多苷、雷公藤甲素等雷公藤的制剂[2]。雷公藤多苷是从雷公藤根中提取精制而成的一种脂溶性混合物,为我国首先研究利用的抗炎免疫调节中草药,具有“中草药激素”之称[3]。临床上可用于治疗类风湿性关节炎、原发性肾小球肾病、肾病综合征、紫瘢性及狼疮性肾炎、红斑狼疮、亚急性及慢性重症肝炎、慢性活动性肝炎等免疫系统疾病[4-6]。但是由于雷公藤中的主要有效成分亦为毒性成分,特别是皮部(包括二重皮)毒性较大,超量内服或食入雷公藤嫩芽,或因炮制不当(根皮未剥、原液汁未洗净)等,均可导致人体中毒,甚至死亡[7]。雷公藤中毒症状主要表现为恶心、呕吐、乏力及黑便,严重的会损害中枢神经系统[8-10]。通过对雷公藤毒性、中毒机理及毒性成分进行分析研究发现,引起雷公藤中毒的主要成分是二萜内酯类,其次为生物碱类,主要包括雷公藤甲素、雷公藤内酯甲、雷公藤晋碱及雷公藤次碱[11]。
目前国内外针对雷公藤中毒与毒性成分的研究主要集中在雷公藤药材中各成分的检测[12-13],以及分析雷公藤中活性成分新的药效研究[14]。常用的分析方法主要有比色法[15]、高效液相色谱法[16]。上述方法主要用于雷公藤药材及制剂中各成分的测定,但用于雷公藤中毒者呕吐物、血液、尿液等中毒样品的测定则存在检测时间长、检测灵敏度低等缺点。钟世豪[17]采用QuEChERS-HPLC-MS/MS 法对生物样品如血液、肝脏、尿液中包括雷公藤吉碱、雷公藤次碱、雷公藤春碱、雷公藤定碱等在内的24种植物毒素进行了测定,但因样品性质不同,此方法不适用于中毒者呕吐物的测定。笔者针对雷公藤急性中毒者的呕吐物,建立一种快速、准确的超高效液相色谱-串联质谱检测方法,对中毒患者抢救及中毒案件分析具有理论和现实意义。
1 实验部分
1.1 主要仪器与试剂
超高效液相色谱-串联质谱仪:Acquity Xevo TQ 型,美国沃特世公司。
高速冷冻离心机:Thermo ST16R 型,美国赛默飞世尔科技公司。
漩涡混合器:Vortex-genie2 型,美国Scientific industries 公司。
分析天平:BS 110S 型,感量为0.1 mg,德国赛多利斯公司。
雷公藤次碱、雷公藤晋碱、雷公藤甲素、雷公藤内酯甲标准品:纯度(质量分数)均不小于98%,批号分别为20113、20625、20209、20907,南通飞宇生物科技有限公司。
雷公藤中毒者胃内容物冻干粉样品:简称冻干粉样品,编号为ZW 054,由中国疾病预防控制中心职业卫生与中毒控制所提供。
胃内容物冻干粉阴性样品:编号为ZW051,简称冻干粉阴性样品,由中国疾病预防控制中心职业卫生与中毒控制所提供。
乙腈、甲醇:均为色谱纯,美国赛默飞世尔科技公司。
甲酸铵、甲酸:均为优级纯,北京化工厂。实验用水为超纯水。
1.2 溶液配制
标准储备液:称取雷公藤次碱、雷公藤晋碱、雷公藤甲素和雷公藤内酯甲标准品各(2.5±0.1)mg,分别置于4 只10 mL 容量瓶中,分别加入甲醇溶解并定容至标线,配制成雷公藤次碱、雷公藤晋碱、雷公藤甲素、雷公藤内酯甲的质量浓度分别为0.240 0、0.260 0、0.255 0、0.249 6 mg/mL 的标准储备液,于-20 ℃保存。
标准中间液:准确吸取上述雷公藤次碱、雷公藤晋碱、雷公藤甲素、雷公藤内酯甲标准储备液各0.2 mL,分别置于4 只10 mL 容量瓶中,分别加入甲醇稀释定容至标线,配制成雷公藤次碱、雷公藤晋碱、雷公藤甲素、雷公藤内酯甲的质量浓度分别为4.8、5.2、5.1、5.0 μg/mL 的标准中间液,于-20 ℃保存。
混合标准工作液:依次准确吸取雷公藤次碱、雷公藤晋碱、雷公藤甲素、雷公藤内酯甲标准中间液1.0、1.0、1.0、2.0 mL,置于同一只10 mL 容量瓶中,加入10%甲醇-0.1%甲酸溶液定容至标线,配制成雷公藤次碱、雷公藤晋碱、雷公藤甲素、雷公藤内酯甲的质量浓度分别为0.48、0.52、0.51、1.0 μg/mL的混合标准工作液,于-20 ℃保存。
系列混合标准工作液:依次准确吸取雷公藤次碱、雷公藤晋碱、雷公藤甲素、雷公藤内酯甲混合标准工作液0.05、0.1、0.2、0.5、1.0 mL,分别置于5 只1 mL 容量瓶中,分别加入10%甲醇-0.1%甲酸溶液定容至标线,配制成雷公藤次碱、雷公藤晋碱、雷公藤甲素、雷公藤内酯甲系列混合标准工作液,-20℃避光保存。系列混合标准工作液中雷公藤次碱、雷公藤晋碱、雷公藤甲素、雷公藤内酯甲的质量浓度见表1。
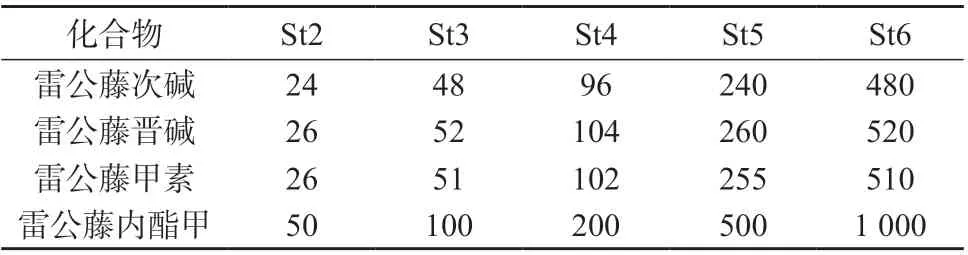
表1 系列混合标准工作液质量浓度 ng/mL
1.3 仪器工作条件
1.3.1 色谱
色 谱 柱:Waters ACQUITY UPLC BEH C18柱(100 mm×2.1 mm,粒径为1.7 μm,美国沃特世公司);柱温:40 ℃;样品室温度:4 ℃;流动相:A 相为乙腈;B 相为10 mmol/L 甲酸铵-0.1%甲酸溶液;洗脱方式:梯度洗脱;洗脱程序:初始A 相体积分数为5%,0~3 min,A 相体积分数由5%逐渐上升至95%并保持至第7 min,7.01 min 开始A 相体积分数降回5%并保持至第8 min;流量:0.3 mL/min;进样体积:10 μL。
1.3.2 串联质谱
离子源:电喷雾离子源,正离子扫描模式;毛细管电压:2.64 kV;离子源温度:150 ℃;锥孔反吹气:氮气,纯度(体积分数)大于99.999%,流量为50 L/h;脱溶剂气:氮气,温度为350 ℃,流量为650 L/h;采集方式:多反应监测(MRM)模式。4 种毒性化合物质谱参数及保留时间见表2。
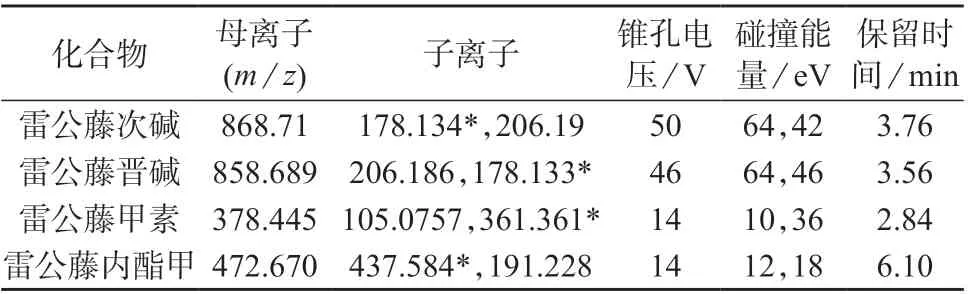
表2 4 种毒性化合物的质谱参数及保留时间
1.4 样品处理
准确称取0.1 g(精确至0.001 g)冻干粉样品,置于15 mL 离心管中,加入10 mL 甲醇,超声提取60 min,以10 000 r/min 转速离心10 min,取上清液与水按体积比为1∶1 稀释,过0.22 μm 滤膜后进样测定。
2 结果与讨论
2.1 色谱柱选择
4 种待测物均为脂溶性化合物,适用于反相色谱柱对其进行分离检测,分别考察耐100%水相、对极性基质干扰有较好分离效果的Waters ACQUITY UPLC HSS T3 柱和对中等极性和弱极性化合物均有较好保留的通用型Waters ACQUITY UPLC BEH C18柱对4 种待测化合物的分离效果。结果显示,两种色谱柱均能实现对待测化合物的保留,但BEH C18柱保留时间更为适宜,尤其是针对雷公藤内酯甲,可将出峰时间提前3 min,单个样品测定时间缩短至8 min,因此选择Waters ACQUITY UPLC BEH C18柱作为样品分析柱。
2.2 流动相选择
超高效液相色谱-串联质谱仪常用的有机相一般为甲醇或者乙腈,当待测物的灵敏度极低时,以甲醇和乙腈的混合液作为有机相,可以提高待测物响应值。针对雷公藤晋碱、雷公藤次碱、雷公藤甲素和雷公藤内酯甲的测定,分别考察了甲醇和乙腈作为有机相时的测定结果,表明乙腈作为有机相时本底值更低,待测物的灵敏度更高,同时乙腈作为有机相洗脱能力强于甲醇,可以进一步缩短检测时间,因此选择乙腈作为有机相。
在对待测物质谱条件优化时发现,正离子监测模式下,雷公藤甲素和雷公藤内酯甲能够同时监测到(M+NH4+)峰和(M+H+)峰两个母离子,且(M+NH4+)峰的灵敏度远高于(M+H+)峰,因此选择以雷公藤甲素和雷公藤内酯甲的(M+NH4+)峰作为母离子。为了确保(M+NH4+)峰母离子响应值的稳定输出,水相中需添加一定量的甲酸铵和甲酸以提供稳定的铵离子(-NH4+)和氢离子(-H+)环境,经考察对比,最终确定以10 mmol/L 甲酸铵-0.1%甲酸溶液作为水相可得到最佳的色谱峰形及响应值。4 种毒性化合物典型色谱图如图1 所示。

图1 4 种毒性化合物典型色谱图
2.3 提取方法选择
目前对雷公藤药材中脂溶性成分测定时,提取方式多采用甲醇超声提取后经固相萃取柱净化,氮吹浓缩后以高效液相色谱仪测定。此方法步骤复杂耗时,不能满足中毒样品快速测定的要求,同时雷公藤内酯甲的稳定性不佳,氮吹浓缩过程中容易造成损失,影响测定结果。超高效液相色谱-串联质谱仪的灵敏度远高于液相色谱仪,通过对提取净化液进行稀释,降低基质效应和溶剂效应,然后直接进样测定,有助于减少提取过程中待测物质的损失。以空白溶剂加标样品为测定对象,用甲醇超声提取、净化后,分别考察提取净化液经氮吹浓缩后测定和用水按体积比1∶1 稀释后直接测定两种方法的回收率,结果如图2 所示。由图2 可以看出,4 种待测物在40 ℃下经氮气吹干后均造成了不同程度的损失,其中对雷公藤内酯甲损失最大,回收率降至0。对提取净化液稀释后直接进样测定,可以减少提取过程中待测物质的损失,提高测定结果的准确度。

图2 不同提取方法时的回收率
2.4 提取时间选择
称取冻干粉样品0.05 g(精确至0.001 g),以10 mL 甲醇作为提取溶剂进行超声提取,控制超声仪内水温不超过25 ℃,对比不同提取时间时4 种待测化合物的测定结果,结果如图3 所示。
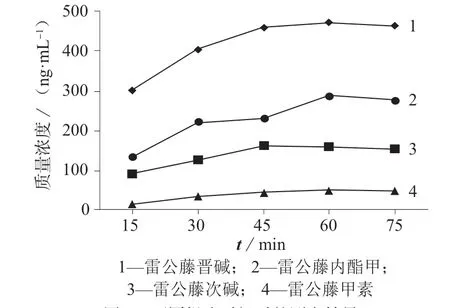
图3 不同提取时间时的测定结果
由图3 可以看出,随着超声提取时间的延长,4种待测物的测定值逐渐增加,当超声提取时间为60 min 时,测定结果达到最大值,超声时间超过60 min后,测定结果略有下降。综合考虑,选择超声提取时间为60 min。
2.4 线性方程与检出限
在1.3 仪器工作条件下,对1.2 中的雷公藤次碱、雷公藤晋碱、雷公藤甲素、雷公藤内酯甲系列混合标准工作液进行测定,以待测化合物的质量浓度为X轴,以色谱峰面积为Y轴,进行回归分析,计算线性方程和相关系数。以3 倍信噪比对应的质量浓度作为方法检出限。4 种毒性化合物的线性范围、线性方程、相关系数及检出限见表3。由表3 可知,4 种毒性化合物在各自的质量浓度范围内与色谱峰面积线性关系良好。

表3 4 种毒性化合物的线性范围、线性方程、相关系数及检出限
2.5 加标回收与精密度试验
准确称取6 份冻干粉阴性样品各0.1 g,分别加入雷公藤晋碱、雷公藤次碱、雷公藤甲素和雷公藤内酯甲标准储备液各25 μL,按1.4 方法处理样品,在1.3 仪器工作条件下分别进行测定,结果见表4。
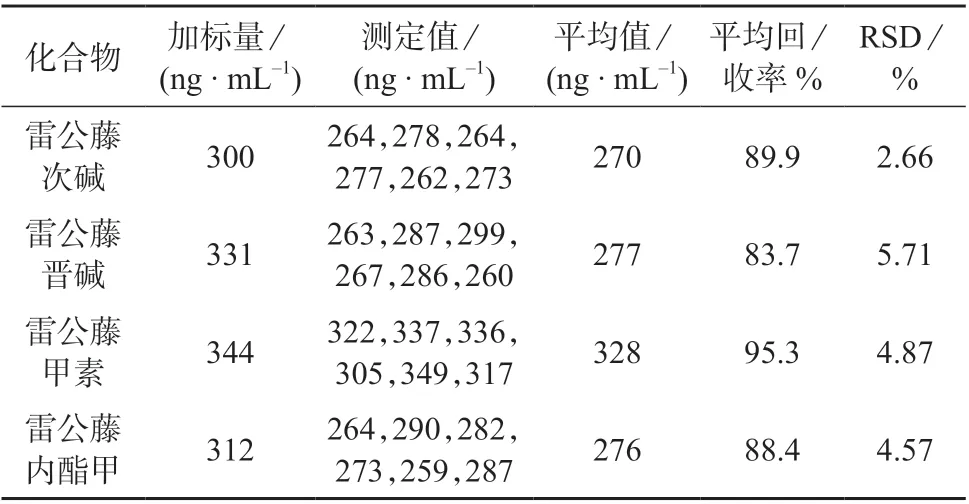
表4 加标回收与精密度试验结果
由表4 可知,4 种毒性化合物的平均回收率为83.7%~95.3%,测定结果的相对标准偏差为2.66%~5.71%,表明该方法的精密度和准确度良好。阴性样品加标色谱图如图4 所示。

图4 阴性样品加标色谱图
3 结语
对急性中毒者胃内容物中4 种毒性成分的测定方法进行了探索,相比于雷公藤药材中脂溶性成分复杂的前处理过程,利用超高效液相色谱-串联质谱仪的高灵敏性,对样品采用甲醇超声提取,稀释后直接上机测定的分析方法,有效缩短了检测时间,提高了检测灵敏度。该方法对于其它类中毒样品检测方法的建立具有重要的参考意义。
猜你喜欢
Journal of Traditional Chinese Medicine(2022年5期)2022-10-14 11:38:06
中华养生保健(2020年9期)2021-01-18 03:11:54
天然产物研究与开发(2018年4期)2018-05-07 06:47:48
制造技术与机床(2017年9期)2017-11-27 02:14:23
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18 10:59:56
中国继续医学教育(2015年2期)2016-01-06 01:36:26
中国病理生理杂志(2015年8期)2015-12-21 12:38:14
西南石油大学学报(自然科学版)(2015年3期)2015-04-16 05:12:08
中国药理学通报(2014年2期)2014-05-09 08:22:23
中成药(2014年11期)2014-02-28 22:29:46
