
Protecting future antimalarials from the trap of resistance: Lessons from artemisinin-based combination therapy (ACT) failures
2021-11-11 13:37NekpenErhunseDinkarSahal
Nekpen Erhunse , Dinkar Sahal
a Malaria Drug Discovery Research Group, International Centre for Genetic Engineering and Biotechnology, New Delhi,110067, India
b Department of Biochemistry, Faculty of Life Sciences, University of Benin, Benin City, Edo-State, Nigeria
Keywords:Artemisinin resistance Quiescence K13 mutations Non-K13 mutations Artemisinin-based combination therapy(ACT) failure Drugs in development Malaria eradication
ABSTRACT Having faced increased clinical treatment failures with dihydroartemisinin-piperaquine (DHA-PPQ),Cambodia swapped the first line artemisinin-based combination therapy (ACT) from DHA-PPQ to artesunate-mefloquine given that parasites resistant to piperaquine are susceptible to mefloquine.However,triple mutants have now emerged,suggesting that drug rotations may not be adequate to keep resistance at bay. There is, therefore, an urgent need for alternative treatment strategies to tackle resistance and prevent its spread. A proper understanding of all contributors to artemisinin resistance may help us identify novel strategies to keep artemisinins effective until new drugs become available for their replacement.This review highlights the role of the key players in artemisinin resistance,the current strategies to deal with it and suggests ways of protecting future antimalarial drugs from bowing to resistance as their predecessors did.
1. Introduction
Malaria continues to be one of the deadliest parasitic infectious diseases of the world. Historically speaking, the use of chemotherapeutic agents has played a remarkable role in the management of malaria [1]. However, their use is fraught with resistance due in part to a high parasite burden as well as the mutability of the parasite's genome [2]. Artemisinins are arguably one of the most successful antimalarial agents to date. Their rapid killing of the parasite coupled with their ability to kill all parasite stages including young ring forms is unmatched[3,4].AlthoughArtemisia annuahas been in use for centuries in the management of malaria in the traditional complementary medicine system of China,it was Youyou Tu and her team who first isolated its active ingredient as artemisinin in 1971[5].Because of its positive impact in combating the disease, she was awarded a joint Nobel Prize in Physiology or Medicine in 2015 (https://www.nobelprize.org/prizes/medicine/2015/summary/). Although artemisinin is demonstrated to be potent against all multi-drug resistant forms ofP.falciparum,some drawbacks to its use are poor oral bioavailability and rapid metabolic clearance.To overcome its low bioavailability,it is used as its semi-synthetic water-soluble (artesunate) and fat-soluble(arteether and artemether) forms which also have better antiplasmodial activity than the parent compound [3]. In a bid to preserve fast acting artemisinin derivatives from resistance, they are administered in combination with longer acting drugs. These partner drugs compensate for the short in vivo half-lives of artemisinins, minimizing the probability of resistance development.While the artemisinins drastically reduce the parasite load in the individual,the few left-over parasites are cleared by the the slowly eliminated partner drug [6]. Besides, since artemisinins will never target the parasite alone, resistance generation will be greatly reduced[6].However,this is not entirely true as artemisinins being faster acting than their partners must be acting alone against early stages of the parasite. The companion drugs for artemisinin in artemisinin-based combination therapies (ACTs) include lumefantrine, amodiaquine, mefloquine, piperaquine and sulfadoxine/pyrimethamine [7] and more recently, pyronaridine [8].
ACTs are currently the first line drugs and have played a significant role in malaria reduction [9] since they were introduced. However,resistance to artemisinin was first reported in Cambodia in 2008[10]and since that time,it has spread to all other countries in the Greater Mekong sub-region [11-13]. Artemisinin resistance is qualitatively different from resistance to other antimalarial drugs like chloroquine.Inthe caseof the formerthere isnochangeintheinvitro IC50between the resistant strain and the sensitive strain[12]while in case of the latter,resistanceisassociatedwithamarkedincreaseinvitroIC50[14].In vivo, artemisinin resistance has been associated with delayed parasite clearance(Supplementary data S1)[15,16],whereas in vitro,it is marked by the ability of resistant parasites to enter a state of quiescence [17] till the persistence time of the drug. Once the drug concentration wanes, the parasite regains its active state of metabolism, leading to the phenomenon of delayed survival [18]. Artemisinin resistance has been described to be mediated by mutations in the Kelch propeller domain of thePlasmodium falciparumK13(pfK13)gene[19,20].Reduced artemisinin efficacy increases the parasite load for the partner drug to handle.This in turn reduces the concentration of partner drug per unit parasite,enhancing the chances of resistance development to partner drug as well as overall treatment failure.This is corroborated by the massive treatment failures in Cambodia found to be related to piperaquine resistance [21-23] associated with amplification of plasmepsins 2 and 3 [24,25]. Further, mefloquine resistance at the Thai-Myanmar border associated with amplification of thePlasmodium falciparummultidrug resistance gene-1(PfMDR1) reappeared soon after artemisinin resistance was established[26].Both these biological responses from the drug-challenged parasites have resulted in massive treatment failures. Today, ACT failure is established in the Greater Mekong region of Cambodia[11-13]and is recognized as a decreased susceptibility to artemisinin as well as resistance to the partner drugs[21,22].
The Greater Mekong sub-region is regarded as the epicentre for drug resistance because resistance to all antimalarial drugs including ACTs has begun from this region [27] from where it has spread to other regions of the world. Thus, resistance to chloroquine and sulphadoxine/pyrimethamine had earlier started from this region from where it had spread to India and then to East Africa and eventually all of Africa [27]. This is a reminder of the fact that millions of lives were lost when chloroquine resistance emerged in sub-Saharan Africa between the 1980s and 1990s [28]. Recently,two non-synonymous mutations in the propeller region of K13 associated with delayed parasite clearance in patients on ACTs and corresponding to high rate of parasites survival in a ring-survival assay (RSA) were identified in Changlang district of Arunachal Pradesh, Eastern India [29]. More worrying is the recent report of the de novo emergence of artemisinin resistant K13 R561H mutant parasites in Rwanda [30]. Should history repeat itself, and artemisinin resistance spread to all of Africa, it could be disastrous[31-33]. Therefore, novel fast acting drugs with novel targets on the parasite are urgently required.Further,a better understanding of the molecular mechanisms behind artemisinin resistance may provide the right strategies to preserve them until the new agents are ready to replace them. Currently, the norm is the rotation of ACTs whereby the ACT which has become ineffective in a region is swapped with another and evaluated after every 2 years [27].However, the emergence of triple mutants in Cambodia [34] suggests that better strategies are urgently needed. This review highlights the molecular mechanism of resistance to artemisinin and current strategies to contain artemisinin resistance, and discusses ways to better protect future drugs from becoming prey to resistance.
2. Resistance to artemisinin and its partners in artemisinin combination therapies (ACTs)
Several non-synonymous mutations in propeller region of K13[35](Fig.1)have been linked to decreased susceptibility of parasites to artemisinin.SuchPfK13 mutations are responsible for creating a scenario where the partner drugs are confronted with more parasite load than they can deal with. This causes partner drug resistance, resulting in ACT failure [21,22]. In a surveillance study, a single fit artemisinin resistant lineage (C580Y) was reported to acquire piperaquine resistance and spread to Thailand and Laos from Cambodia outcompeting other K13 mutants [36].
A different scenario, however, occurred in high transmission areas where due to the high frequency of infections, a higher density of parasite was exposed to the long half-life drug partners of artemisinin [37]. This places partner drugs at risk of resistance even in the absence of decreased parasite susceptibility to artemisinins [38]. Indeed, while several K13 mutations have been reported in Africa, none of them has been linked to artemisinin resistance [39-41] except for a very recent report in Rwanda[30].While this mutation has not led to widespread treatment failures[30], resistance conferring mutations to artemisinin partner drugs has been observed to result in reduced efficacy of ACTs in Africa[39,42].
Two digestive vacuole (DV) transporters:PfMDR1 whose gene product is the P-glycoprotein homologue-1 (Pgh-1) andP. falciparumchloroquine resistance transporter (PfCRT) play major roles in partner drug resistance [43-45].PfMDR1 is a member of the ATP-binding cassette superfamily of transporters and a homologue of the human P-glycoprotein [43,46]. It is mainly expressed on the surface of the DV, where it mediates the transport of physiological solutes as well as several antimalarials into the DV [47-51]. A schematic representation of the topology ofPfMDR1 on the parasite's DV [52-54] together with other DV transporters [55] and their inhibitors [46,52,56-59] is presented in Fig. 2.
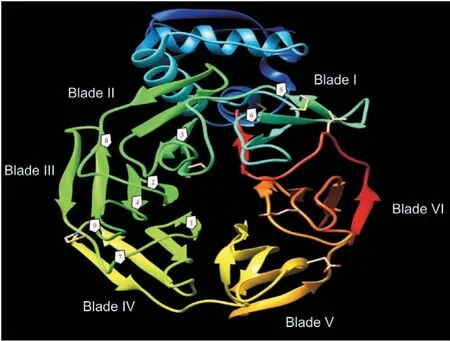
Fig.1.Kelch 13 (K13) mutations implicated in decreased efficacy of artemisinins. The propeller domain of the kelch protein possesses six blades (or kelch) repeats. The frequent mutations reported in Cambodia, Laos, PDR, and Vietnam are depicted as numbers 1-4 while 5-8 represent the frequent mutations in China, Myanmar, and Thailand. The kelch mutation 9 is found in all countries of the Greater Mekong subregion [35]. 1: C580Y; 2: R539T; 3: Y493H; 4: I543T; 5: F446L; 6: N458Y; 7: P574L;8:R561H;9:P553L.The predicted 3D structure of the protein with PDB number 4YY8 was visualized using the Chimera software, version 1.13.1.

Fig.2.The pleiotropic digestive vacuole (DV) transporter PfCRT carries mutations conferring resistance to various antimalarial drugs including artemisinin partner drugs. Mutations and/or copy number variations in PfMDR1 (Pgh-1) have also been observed to result in partner drug resistance (See Table 1). Pgh-1 is the parasite's homologue of the human P-glycoprotein which it mainly expresses on the surface of its DV.During DV formation,the topology of Pgh-1 is inverted resulting in the transport of substrates into the parasite's DV[46,52].Pgh-1 transports many antimalarial drugs into the DV. PfCRT, on the other hand, transports many of these antimalarial drugs out of the DV. The DV's acidic pH is thought to play a critical role in its various functions.Acidification is done by the V-type proton-ATPase (H+-ATPase) as well as a protonpyrophosphatase (H+-PPase) [55]. The activity of these DV transporters can be inhibited by various compounds as indicated. ONT-093 and XR-9576 (Tariquidar) are 3rd generation P-glycoprotein inhibitors[56]which have been demonstrated to inhibit Pgh-1 [46,52]while verapamil partially reverses resistance to chloroquine and related compounds by inhibiting PfCRT [57-59].
Although mutations and copy number variation (CNV) inPfMDR1 gene affect the in vitro response to artemisinin, their utility as biomarkers for artemisinin resistance in the field has,however, proven to be of limited use [45]. Its involvement in resistance of artemisinin partner drugs is however established[60]. Unlike thePfMDR1,PfCRT transports solutes out of the DV.Together, both transporters have played huge roles in the loss of many antimalarials, notably chloroquine, quinine, amodiaquine and piperaquine, to resistance. Their location on the surface of the DV allows them to modulate the activity of these drugs by influencing their concentrations at the site of action [44,52].Interestingly, ACT partner drugs, (a) amodiaquine and lumefantrine and (b) piperaquine and mefloquine, cause opposing selective pressure (Supplementary data S2) onPfMDR1 andPfCRT haplotypes [61-63]. This constitutes the basis for swapping of one failed ACT with a new ACT [27,34] as well as the ideas behind triple ACTs [64]. Table 1 provides the mechanisms of action (MoA) of artemisinins and their partners in ACTs[65-73] as well as their molecular markers of resistance[19,49,50,74-91].

Table 1 Mechanism of antiplasmodial action,year of introduction,first reports of resistance,molecular markers for resistance and resistance mechanisms of artemisinins,and partner drugs.
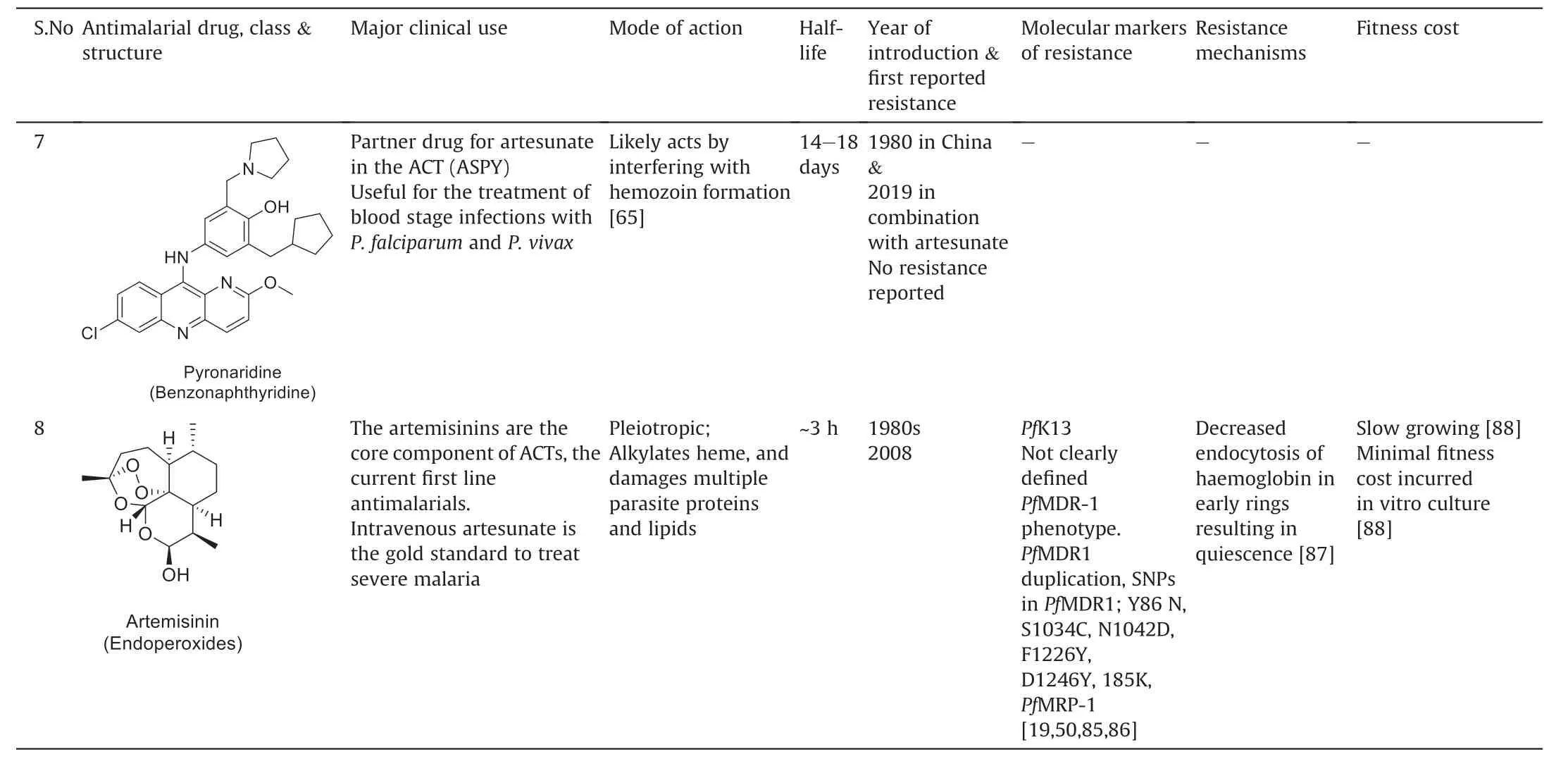
Table 1 (continued)
2.1. Molecular mechanisms of artemisinin action and resistance
Resistance to artemisinin is evidenced by delayed clearance of the parasite due to decreased susceptibility of its young ring forms.The induction of developmental arrest of a small (<1%) subpopulation of rings (0-3 h post-invasion) (Fig. S1) which enter a quiescence state together with the killing of other stages cause parasites to score susceptible using the standard in vitro susceptibility assays. Consequently, the ring stage survival (RSA) assay(Supplementary data S3) which entails a short exposure to the pharmacologically relevant dose of dihydroartemisinin followed by drug withdrawal is the golden standard for investigating artemisinin resistance [92].
Artemisinins are activated by iron-bound heme monomers which are degradation products of hemoglobin. Heme iron activates artemisinin due to the cleavage of the endoperoxide bond generating free radicals via a Fenton mechanism which lead to the formation of hemarts (heme-artemisinin) which suppress heme detoxification [93] and damage cellular macromolecules including parasite membrane components, proteins and lipids[94,95].
Clinical resistance to artemisinin was first detected and reported in Cambodia in 2006 and 2008, respectively [7]. Mutations in the BTB/POZ or propeller domain of the K13 gene (Pf3D7_1343700)which is located on chromosome 13 have been associated with artemisinin resistance [19,20]. With just one mutation enough to cause resistance [20], all mutations found in this gene correlating with artemisinin resistance are non-synonymous [35] (Fig. 1).Further, they are located after amino acid residue 440 of the 726 residues long K13 protein [19,20,41]. Except for the association to artemisinin resistance, the role of K13 in the parasite remained obscure for a very long time [18,96] and has only recently been deciphered [87,97]. There appear to be some conflicts as to the localization of K13 within the parasite, with its presence observed in a doughnut-like compartment within cytostome-like organelles at the parasites periphery [96] versus in a compartment close to cytostome-like organelles in close proximity with the DV [87].However, both groups have provided a strong link between an impaired hemoglobin catabolism and artemisinin resistance[87,96]. More particularly, it was Birnbaum et al. [87] who discovered the relationship between decreased hemoglobin endocytosis and artemisinin resistance. They described K13 and 10 of its 12 interacting protein candidates(K1Cs)to all localize in a subcellular compartment termed the K13 compartment. A non-K13 protein(ubiquitin carboxyl-terminal hydroxylase1 (UBP1)) has previously been reported to be mutated in rodent malaria parasites resistant to artesunate [98]. Using a previously established bloated food vacuole phenotype assay[99],Birnbaum et al.[87]showed that 4 of the K13 compartment proteins including UBP1, the parasite's homologue of the endocytosis protein Eps15, KIC7 and Ap-2μ were required for the endocytosis of hemoglobin into the DV in all asexual blood stages of the parasite while K13 was shown to be needed for this process only in rings. This agreed strongly with their previous finding that only ring stage parasites are affected by the inactivation of K13[100].Furthermore,mutations in either K13 or 8 of the 12 KICs were reported to result in resistance to artemisinin.This observation coupled with the fact that certain mutant parasites lacking K13 mutations have previously been observed to display the increased ring-stage survival phenotype [101,102]typical of artemisinin resistance made the authors to probe the possibility of a mutation in one of the K13 interacting proteins.Indeed, R3138H mutation in UBP1 had earlier been identified by a genomic surveillance of parasite field samples to confer resistance to artemisinin [101]. Interestingly, Birnbaum et al. [87] have now shown this non-K13 mutation causes resistance to artemisinin.
Taken together, since hemoglobin is required as a source of amino acids for the parasite's growth, its decreased uptake due to mutations in the kelch gene causes the parasites to grow at a slower rate,a phenotype which appears to be a biological tradeoff[87,97].The parasite thus utilizes this enhanced quiescence capacity/decreased growth rate to prepare itself to cope with the adverse environment, which enables it to resume growth when the drug has been cleared. This is evidenced by the increased constitutive expression of the unfolded protein response(UPR)pathway in the quiescent state conferring these resistant parasites an increased ability to withstand the cellular stress caused by artemisinin toxicity [18].
2.2. Current strategies for combating resistance to ACTs
To manage ACT resistance in regions where failure to one ACT is established,the norm has been to swap the failed ACT with a new ACT [34]. This idea of swapping has its origin in the opposing selective pressures between (a) amodiaquine and lumefantrine and (b) piperaquine and mefloquine onPfMDR1 andPfCRT haplotypes(Supplementary data S2)[61-63].Recently,the safety and efficacy of triple ACTs which include artemisinin and two partner drugs with opposing drug pressure selection have been investigated (NCT02453308) [103]. In Vietnam, mefloquine has already been added to the ACT of dihydroartemisinin-piperaquine[27,104]. Artemether-lumefantrine (co-artemether) and artesunate-amodiaquine are the ACTs mainly used in Africa[105].A recent survey revealed a reduced prevalence of thePfMDR1 haplotypes (86Y and 1246Y) associated with lumefantrine resistance in African countries where co-artemether has been in use for about 10 years [42]. This is consistent with a decreased sensitivity to co-artemether and an increased sensitivity to artesunate-amodiaquine. In the event of treatment failures,retreatment with an alternative ACT (i.e., treating co-artemether failure with artesunate-amodiaquine and vice versa) has been suggested [106]. Further, it may be more prudent to introduce triple ACTs combining co-artemether with amodiaquine as a stopgap [27,107] in Africa so as to avert avoidable loss of lives as previously recorded before the endorsement of ACTs [108].However,these stopgap strategies have been suggested to be only short or mid-term measures to manage the situation [27,37,109].Indeed, long-term strategies will have to be found to sustain the therapeutic lifespan of the artemisinins until other options are available for their replacement as triple mutants have appeared in Cambodia after dihydroartemisinin-piperaquine was swapped with artesunate-mefloquine [34]. Even though this has not resulted in widespread treatment failures [34], it may, however,just be a matter of time before resistance is established and clinical failures are recorded. Other strategies that have been suggested to manage artemisinin resistance and prevent its spread include adding antigametocytocidal drugs to the ACTs to be used in regions where resistance is established [12], extending the duration of the 3-day course of ACTs [110-112], increasing the dose of the partner drugs [113], utilizing multiple first-line antimalarials [42], developing nano-based formulations[114-116],replacing artemisinin altogether with novel drugs that are fast-acting [117-120] and ACTs proteasome inhibitors which act as an artemisinin activation booster [121,122].
In line with replacing the artemisinins,several drugs(Fig.3)are currently in different stages of development.However,before they can be made available,they will have to pass obligatory safety and efficacy tests. Further, their partners in combinations will have to be determined [27,123].
2.3. Antimalarial drugs in development for the treatment of malaria
Several candidate drugs(Fig.3)with good pharmacological profiles are currently in different stages of development[124].However,drug development is a very lengthy(Fig.4)[124]and cost intensive process [125]. Therefore, to manage the situation, the ACTs which have been renderedineffective havebeen swappedwhile thesafetyof triple ACTs, comprising a standard dual ACT with another slowly eliminated antimalarial, has been evaluated for use as stopgaps[103,109,124].Combinations of dihydroartemisinin+piperaquine+mefloquine and artemether + lumefantrine + amodiaquine have successfully completed clinical trials (ClinicalTrials.gov identifier numbers NCT02612545 and NCT02696954 respectively) and were foundtobeefficacious(eveninthefaceofartemisininresistance),safe and well tolerated[103].
Another triple ACT undergoing clinical trials uses imatinib, an anticancer drug repurposed for the management of malaria which is currently in phase II trials (NCT036976 68) as a triple combination with dihydroartemisinin-piperaquine [1]. Together with their chemical structures(Fig.3),the profiless of some of the drugs in development for the treatment of malaria are given below below.
2.3.1. Cipargamin (KAE609)
Cipargamin, a fast and long-lasting spiroindolone [117,124,126]that acts by inhibiting P-type transporter Na+-ATPase (PfATP4)[127], was discovered by a partnership among Novartis, the STPHI and the Wellcome Trust [118,128]. With potency comparable to artesunate, it is equipotent against drug-resistant strains ofP. falciparumandP. vivaxisolates [119,129]. However, combination with a longer acting partner drug without cross resistance toPfATP4 is suggested [124]. Currently, it is in phase IIb of the drug development pipeline (NCT03334747).
2.3.2. (+)-SJ733
(+)-SJ733 is a dihydroisoquinolone which like cipargamin is also an inhibitor of thePfATP4. Discovered by a partnership between St Jude Children's Research Hospital and Rutgers University in 2010,(+)-SJ733 is a blood stage schizonticide forP.falciparumandP.vivaxand also inhibits gametocytogenesis [130,131]. Further, it possesses an excellent anti-malarial activity in vivo[131].Currently in phase 1a of development, the first human study for this drug was a singledose escalation study completed in 2018 after which a three-dose cohort study is to be undertaken(NCT02661373).
2.3.3. Artefenomel-ferroquine
Artefenomel-ferroquine is a combination of a fast- and longlasting (elimination half-life of 46-62 h) synthetic ozonide, artefenomel (OZ439) [132,133], and ferroquine (FQ), a 4-aminoquinoline which retains activity against chloroquine- and piperaquineresistant parasites in vitro with a long elimination half-life of 16 days.The replacement of the alkyl group in arteferolane(OZ377,t1/2=24 h) with a phenyl group led to the synthesis of the 2nd generation ozonide artefenomel(OZ439)with a significantly increasedt1/2of 46-62 h[132].Further,it was interesting that the increment int1/2was not at the cost of reduced reactivity of iron with the peroxide.The mechanism of action of artefenomel like other peroxidecontaining antimalarials is not precisely defined although it is reported to involve oxidative stress [134,135]. An advantage of this combination of artefenomel and ferroquine is that neither of the constituent drugs has previously been deployed as monotherapy.Although the ozonide did not show any cross resistance for parasites carrying the well-known K13 (C580Y) mutation, there was cross resistance for parasites carrying the K13 (I543T) mutation [136].However, the goal to create a single-dose curative treatment with this combination in a dose-finding phase IIb study was recently terminated in 2019 as certain criteria were not met(NCT02497612).
2.3.4. Lumefantrine-KAF156
Lumefantrine-KAF156 combines the aryl-amino alcohol (lumefantrine) with the highly potent imidazolopiperazine KAF156, a fast-acting drug with multistage activity and an elimination halflife of 48.7 ± 7.9 h [137]. KAF156 was identified in 2008 by Novartis and the Scripps Research Institute [138]. Although the MoA for KAF156 remains unknown, mutations generated under KAF156 pressure have been identified in three gene targets of the parasite:P. falciparumcyclic amine resistance locus (PfCARL) and transporters of UDP-galactose and acetyl-CoA respectively [139],suggesting a novel MoA for the drug.KAF156 is currently in phase IIb clinical trials, where dose combinations with lumefantrine are to be explored(NCT03167242).
2.3.5. Fosmidomycin-piperaquine
Fosmidomycin was developed as an antibacterial drug in the 1980s.However,as a result of drug repurposing[1],it is currently indevelopment as an antimalarial.It has a good safety profile and acts by inhibiting the parasite's isoprenoid biosynthetic pathway[140].Most recently,it was partnered with piperaquine,in a phase II trial.The status of its progress is, however, currently unknown(NCT02198807).
2.3.6. AQ-13
AQ-13 is a chloroquine (CQ) derivative first described in 1946[141]. It has CQ-like MoA and pharmacokinetic properties. Unlike CQ which has a methyl harbouring 4-carbon chain,AQ-13 with a 3-carbon desmethyl chain is equipotent against both CQ sensitive and CQ resistant strains of the parasite [142]. It has been in clinical development for over 10 years.It last completed phase II trial at the end of 2017 (NCT01614964) with the study revealing that it is not inferior to artemether-lumefantrine [143]; however, the status of this compound is currently unclear as there has been no mention of any follow-up trials since that time.
2.3.7. MMV 390048
Identified in 2012 by a team at the University of Cape Town,South Africa [144], MMV 390048 (3-(2-(Trifluoromethyl) pyridin-5-yl)-5-(4-(methylsulfonyl)-phenyl) pyridin-2-amine) is a multistage antimalarial that acts by inhibitingP. falciparumphosphatidylinositol 4-kinase [145]. This aminopyridine possesses good prophylactic activity againstP. cynomolgiin vivo and has the potential to act as a transmission-blocking drug[144].It is currently in phase IIa trial in a proof-of-concept human study to confirm its observed activity in pre-clinical trials (NCT02880241).
2.3.8. DMS265
DMS265 (2-(1,1-difluoroethyl)-5-methyl-N-[4-(pentafluoro-λ6-sulfanyl) phenyl]-[1,2,4] triazolo[1,5-α] pyrimidin-7-amine) belongs to a class of inhibitors of the parasite's dihydroorotate dehydrogenase (DHODH) enzyme required for pyrimidine biosynthesis with potency against both blood and liver stages [146] ofP.falciparum.It has an excellent safety profile and an exceptionally low clearance rate in humans[147].DMS265 has completed a phase IIa trial in Peru in patients withP. falciparum/P. vivaxinfections(NCT02123290).While the single dose study with an 80%cure rate was successful forP.falciparuminfection,no cure was recorded forP.vivaxinfection.This pattern is reminiscent of the only single dose clinically deployed antimalarial sulphadoxine-pyrimethamine which demonstrated high efficacy of a single dose againstP. falciparum, but not against againstP. vivax[148]. Single point mutations in the dhodh gene were, however, seen in patients withP.falciparuminfection recurrence, suggesting that DMS265 readily selects for resistant parasites. Also, theP. falciparumD10 (CQ-sensitive)strain of the parasite which has previously been observed to be less susceptible to DHODH inhibitors was also found to be resistant to DMS265 [147,149]. Its relatively low barrier of resistance coupled with the fact that in comparison with the newer antimalarials it is a less potent inhibitor of the parasite suggests that its deployment in combination with a more potent agent may be necessary[147,148,150].In this regard,DMS265 has completed a controlled human malaria infection study in combination with OZ439 in a phase Ib dose finding study to characterize the safety of the combination and pharmacokinetic and pharmacodynamic interactions between both drugs (This study has been registered at ClinicalTrials.gov under identifier NCT02389348) with the results from the study supporting further clinical development of the combination [151].
2.3.9. M5717 (DDD 107498)

Fig.3.Chemical structures of some candidate drugs in development for the treatment of malaria.The TCs each fitting into under SERCaP is indicated in red at the bottom of each box.
M5717 (6-Fluoro-2-[4-(morpholin-4-ylmethyl) phenyl]-N-(2-pyrrolidin-1-ylethyl) quinoline-4-carboxamide), formerly known as DDD 107498, is an inhibitor of translation elongation factor 2(eEf2) responsible for the GTP-dependent translocation of the ribosome along messenger RNA-a step essential for protein synthesis [152]. M5717 is a multiple stage antimalarial with activity against pre-erythrocytic stages, blood stages, as well as male and female mature gametocyte forms of the parasite[152].The first inhuman study for this drug was recently completed(NCT03261401).However, the findings have not been made known.
2.3.10. ACT-451840
ACT-451840 ((S,E)-N-(4-(4-acetylpiperazin-1-yl) benzyl)-3-(4-(tert-butyl) phenyl)-N-(1-(4-(4-cyanobenzyl) piperazin-1-yl)-1-oxo-3-phenylpropan-2-yl) acrylamide) is a compound with a novel mode of action that was developed in 2016 through a collaboration between Actelion Pharmaceuticals and the Swiss Tropical and Public Health Institute (STPHI) [120]. It is a potent inhibitor of both multidrug-resistant and drug-sensitiveP.falciparumasexual blood stage parasites[153]and is a fast-acting compound with a long half-life acting against all blood stages ofP.falciparumincluding stage V gametocyte [120].
The lead compound ACT-213615 [154] which was optimized to ACT-451840[1]has been shown to interact withPfMDR1[154],and mutations onPfMDR1 by ACT-451840 treatment also resulted in decreased efficacy for ACT-213615. Hence, ACT-451840 is also thought to act by directly targetingPfMDR1 [153].
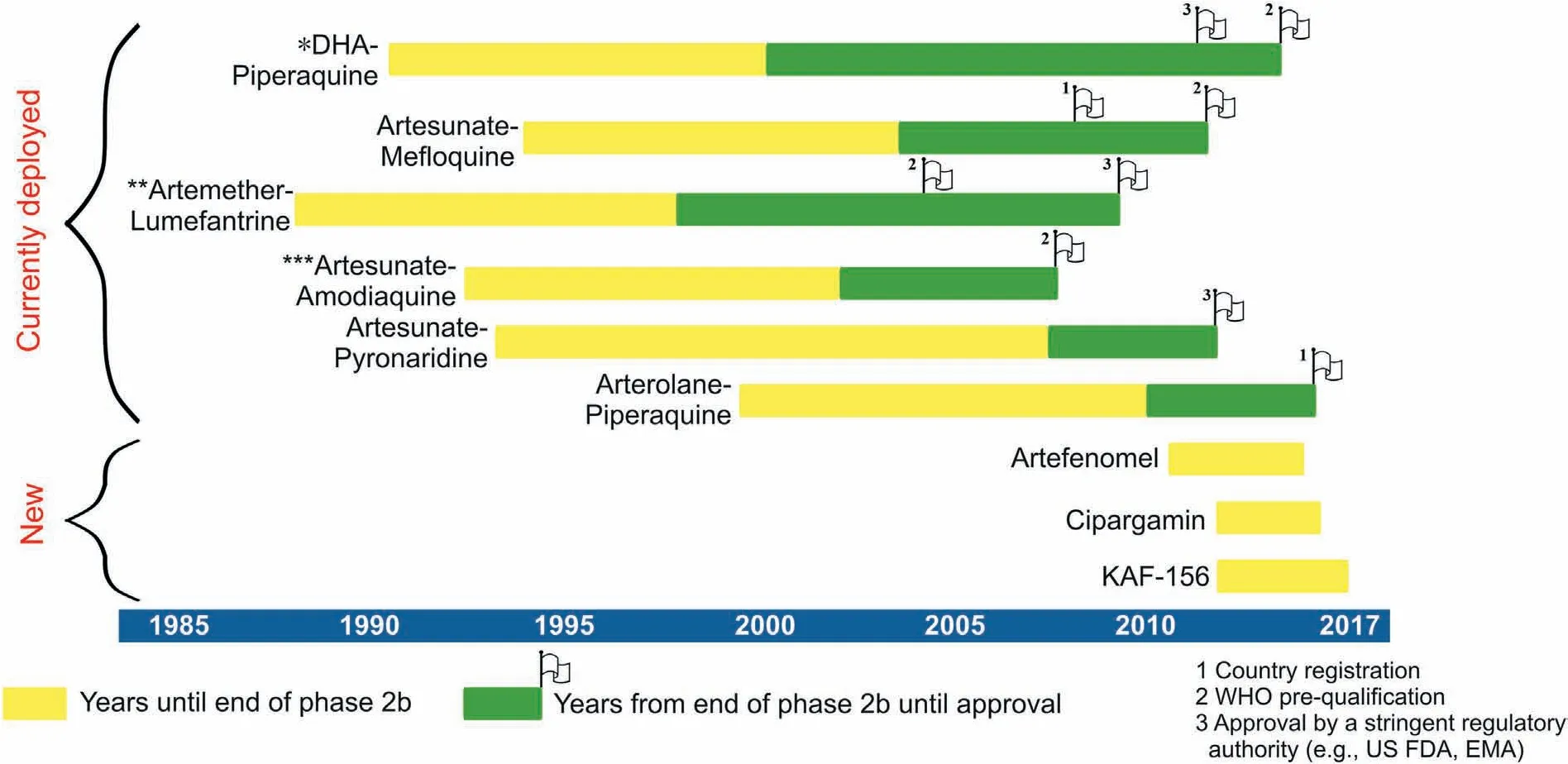
Fig.4.Developmental timeline of currently deployed ACTs versus leading antimalarial compounds for the treatment of malaria. Single asterisk: Dihydroartemisinin-piperaquine(WHO pre-qualification refers to eurartesim by sigma tau in 2015) approved by EMA in 2011. Double asterisk: Artemether-lumefantrine (WHO pre-qualification for coartem by Novartis in 2004,products by Ipca and Cipla in 2009;others have followed since then).Triple asterisk:Artesunate-amodiaquine(WHO pre-qualification for Sanofi's ASAQ Winthrop in 2008, Ipca and Guilin products in 2012, Ajanta in 2013 and Cipla in 2014). Adapted from Ref. [124], http://creativecommons.org/licenses/by-nc/4.0/.
Intriguingly, mutations conferred onPfMDR1 following ACT-451840 exposure resulted in the parasite becoming more susceptible to several clinically deployed antimalarials including artemisinins,as well as its partners in ACTs,viz.lumefantrine,mefloquine,monodesethyl amodiaquine, halofantrine and piperaquine [153].This observation is remarkable as this opposing selective pressure may be therapeutically beneficial. A similar observation was reported by Vanaerschot et al. [155] who screened a hexahydroquinoline (HHQ) series of antimalarials from a Novartis-GNF Malaria Box. These researchers discovered three potent inhibitors of the parasite which inhibited the import of haemoglobin into the parasite's DV [155,156]. Mutations associated with HHQ resistance also resulted in increased susceptibility to first-line antimalarials(lumefantrine, dihydroartemisinin, piperaquine, mefloquine, and quinine). Interestingly, these HHQ mutant parasites showed cross resistance to ACT-451840. The researchers thus reported that the HHQs inhibitPfMDR1. Like ACT-451840, HHQs are also fast acting against all erythrocytic stages of the parasite including the early and late gametocyte stages. HHQs, however, require some optimisation before they can be considered in the developmental pipeline as they are not very potent in vivo [155].
Given the role of Pgh-1 in the resistance to several antimalarials[47-51],targeting this transporter with drugs like ACT-451840 may be rewarding. Furthermore, ACT-451840 inactivates stage V gametocytes which are refractory to many antimalarials [157]. With such a profile,ACT-451840 has a great potential of being considered as both a TCP 1 and TCP 3b (Table 2 [158,159]) drug candidate. Its first in-human study was completed in 2014 (ClinicalTrials.gov,identifier number NCT02223871) and is currently awaiting a decision to proceed [160].
3. Lessons learnt from the failure of ACTs
Driven by the need to prevent treatment failures, the logic for combining artemisinin with longer acting drugs in ACTs is to enable artemisinin to do the rapid killing of a majority of the parasites and make the long-lasting slower acting partner drug kill the remaining few parasites [161]. It is also suggested that the rapid clearance of artemisinin from the circulation will make it very difficult for the parasite to acquire resistance against it. Further, since the parasite would never be exposed to artemisinin alone,resistance generation would be greatly reduced [6]. However, this may not be what obtains as artemisinins being faster acting should target early stages of the parasite alone. Indeed, although it slows to develop artemisinin resistance is now established in all the countries of the Greater Mekong sub-region of Southeastern part of Asia[11,12,162].With the proposed mechanism of artemisinin resistance[87]as the basis and with the eradication goal in view, we discuss our viewpoint on the parasite's resistance to ACTs and propose new therapeutic modalities to protect the novel drugs in development from resistance.
3.1. Complete protection of first-line antimalarials
All antimalarial drugs used for treatment of malaria to date have fallen prey to resistance one after another. Thus, CQ resistance was seen after a decade of successful use [33]. In contrast,resistance to sulfadoxine-pyrimethamine (SP) became evident within months after it was deployed [163]. Thanks to the use of combination strategy, the phenomenon of delayed parasite survival and the resulting treatment failures seen with ACTs took decades [10-12,162]. Recent advances in genomics and proteomics have afforded the targets of most of the drugs in development [127,150,153,164]. This also indicates that resistance can easily be identified in the field. Rapid epidemiological identification of mutations conferring resistance to drugs in use facilitates advance planning for tackling spread of drug resistance.Thus, the presence of existing mutations (e.g., G223R) conferring resistance to thePfATP4 inhibitors (Cipagarmin and +(-)SJ733) in parasite isolates from Africa will affect the development and eventual deployment of this class of compounds [165]. Another clinical candidate DMS265 which has a favorable pharmacokinetic profile can therefore be useful for chemoprophylaxis.Further, like SP, DMS265 can also be given as single dose treatment.However,it easily selects for resistance[150].This suggests a need for DMS265 to be partnered with another drug before deployment. Hence, the development of future antimalarials for use without combining with partner drugs must be deprioritized for formulating the ideal antimalarial single-exposure radical cure and prophylaxis (SERCaP). According to the WHO, SERCaP is likely to be a combination of several chemical entities which fits into different target candidate profiles (TCPs) so as to reduce the risk of resistance. Further, it is intended to be administered as a single dose to improve compliance [166]. The malERA group has spelt out four TCPs dubbed as TCP 1 to TCP 4 under SERCaP(Table 2) which the antimalarial drugs in development must meet [158,159]. But concerns of possible lowered safety margins have been raised since it will mean the exposure of individuals to high concentration of multiple drugs [167]. In spite of the fact that the development of an ideal SERCaP may be very difficult to attain and may take decades, if one is eventually developed, it will contribute immensely to the malaria malaria eradication goal.In the parlance of SERCaP,TCP 1 represents fast acting drugs while the partner TCP 2 represents drugs that could be either fast- or slow-acting but must be bestowed with long half-lives[158]. The artemisinins and their partners in ACTs are thus classical representatives of SERCaPs TCP 1 and 2 drugs, respectively. Artemisinins are the only clinically deployed drugs, which rapidly kill all erythrocytic stages of the parasite including young ring stage.This indicates that even though their partner drugs are present in ACTs, only artemisinin is active against rings [168].Artemisinin resistance is thought to affect parasites at the ring stage because parasite may be more tolerant to toxicity from artemisinin at the relatively inert ring stage [18]. Previously, it was thought that ring stage parasites were the least susceptible to artemisinins as compared to trophozoites and schizonts [169].However, contrary to assumptions, when young rings (2-4 h post-invasion) were observed to be as sensitive to artemisinin as trophozoites and schizonts, they had to be dubbed as “hypersensitive rings” [170,171]. Intriguingly, in artemisinin resistance,a small subset (<1%) of synchronized rings (0-3 h post-invasion)were found to develop a quiescence-based resistance to artemisinin. It is possible that an uneven distribution of cellular effectors [18] among infected cells may be responsible for only a tiny fraction of the ring population displaying this quiescence phenotype [172]. Further, mutations in K13 impairs endocytosis only in rings but not in trophozoites[87].Indeed,among K13 and its interacting partners, K13 is frequently found mutated in resistant parasite isolates [173]. This suggests that the temporal exposure of ring stage parasites to artemisinin alone in ACTs may have played a huge role in ring stage parasites becoming resistant to artemisinin. Thus, placing a slower acting drug in the TCP 2 category may expose the first-line drugs to resistance. It may,therefore,be more rewarding to raise the bar for drugs to be used in the TCP 2 category to include only those drugs which are fastacting and long-lasting. For instance, to avoid future resistancemediated loss of drugs like ACT-451840 (which can fit into the TCP 1 and TCP 3b category) (Table 2), the criterion for selecting its future partners in the TCP 2 category must be only those drugs that are both fast-acting and long-lasting so that at no point can the first-line drugs alone target the parasite. It is important to note that gametocytes from mutants generated by ACT-451840 were demonstrated to be resistant to ACT-451840 [153]. Thissuggests that if resistance to this drug develops in the field, it may be transmissible. Although a fast-acting and long-lasting drug may lead to increased toxicity due to its long half-life, the drug can be optimally tweaked so that a long half-life is not associated with excessive toxicity.

Table 2 Target candidate profiles (TCPs) for SERCaP.
3.2. Partner drugs in combination therapy should be matched judiciously
Artemisinins were paired with their partners not for reasons of synergy or additivity but simply to compensate for their short half-lives so as to minimize the development of parasite resistance. At the first glance, the description of resistance to artemisinin to be the result of impaired endocytosis of hemoglobin [87]would suggest that it may be more beneficial to partner artemisinin with drugs targeting parasite pathways other than those that are downstream of hemoglobin uptake [87,97]. This is because as hemoglobin uptake is compromised by artemisinin resistance,partner drugs which act downstream of hemoglobin uptake such as the quinolines will also be unable to act.However,although the quinoline class antimalarials are thought to act by inhibiting the heme detoxification pathway, increased CNVs of the DV transporter (Pgh-1) actually result in decreased activities for quinine and mefloquine [47]. This serendipitous observation suggests the existence of important cytosolic targets for these antimalarials.Consistent with this, using cell thermal shift assay, Dziekan et al.[68] recently identifiedP. falciparumpurine nucleoside phosphorylase (PfPNP) as a target of quinine and mefloquine. Prior to this, mefloquine had been suggested to inhibit protein translation by binding to the 80S ribosome [73]. Hence, a polypharmacological nature of drugs may explain the synergy reported between artemisinin and its partners [174].
Earlier, the generation of high-grade resistance to artemisinin was investigated using an in vitro recrudescence assay [175].Artemisinin was shown to induce a multidrug tolerance phenotype in the F32-Art5 parasites which were subjected to 5 years of escalating artemisinin pressure. Prolonged exposure of F32-Art5 to artemisinin caused young rings,older rings and trophozoites to show marked resistance to antimalarials of different chemical classes with distinct modes of action including endoperoxides,quinolines and an antifolate, pyrimethamine, but not to atovaquone even when mutations in the antimalarial resistance genes linked to these compounds were absent [175]. Rather, mutations in K13 (M476I), falcipain 2a, PK7, Pfg27 as well as two genes(Pf3D7_1459600 andPf3D7_1464500) of unknown function were observed. In artemisinin resistance, coupled with the suspension of DNA, RNA and protein synthesis, metabolic pathways involved in the synthesis of adenosine triphosphate (ATP) and phosphoenolpyruvate (PEP) are severely downregulated with the parasite depending on the mitochondria and apicoplast for the synthesis of these metabolites to sustain basal metabolism [18,176]. The suspension of RNA and protein synthesis may explain why parasites became resistant to pyrimethamine (which inhibits the folate synthetic pathway) but continued to remain sensitive to atovaquone which targets the mitochondrial electron transport chain[175]. This observation made these researchers suggest the inclusion of atovaquone in ACTs.However,as parasites in Southeast-Asia carry fixed mutations inPfdhps andPfdhfr[177,178],inclusion of atovaquone into ACTs for this region may not be a good option[179].Moreover,the observation by Mˊenard et al.[175]is different from what has been described in the field since after artemisinin treatment failures were established along the Thai-Myanmar border, mefloquine resistance (identifiable by an amplification of thePfMDR1 gene) appeared [26] and piperaquine resistance(identifiable by the overexpression of plasmepsin 2) was seen in Cambodia [21-23]. This seems to suggest an increased parasite burden for partner drugs in the face of resistance to artemisinin.However, unlike K13 mutations which cause inhibition of hemoglobin only in the early ring stage, mutations in K13 interaction proteins prevent hemoglobin uptake in all stages of the parasite[87].This suggests that mutations in these proteins may present a different artemisinin resistance phenotype from K13 mutants.One possibility is that in such a scenario,since artemisinin and the partner drugs require haemoglobin for their action whose uptake would have been compromised at all stages of the parasite,resistant parasites are very likely not to be responsive to artemisinin as well as its partner drugs even in the absence of mutations in genes conferring resistance to the partner drugs. Thus, they may present a similar pluriresistant phenotype like the one observed earlier by Mˊenard et al. [175]. Therefore, the contribution of non-K13 mutations to resistance against artemisinin and its partner drugs needs to be clarified as a matter of urgency to better understand the situation and proffer lasting strategies to manage artemisinin resistance. The emergence of triple mutants(mutation inPfK13(C580Y)as well as amplification ofPfMDR1 and plasmepsin2) following the replacement of dihydroartemisininpiperaquine with artesunate-mefloquine in Thailand, Laos and Vietnam [34] suggests that the opposing drug pressure strategy may fail and that it may be more rewarding to identify drugs like atovaquone that can prevent the parasite's entry into a quiescence state [180] or drugs like theP. falciparumphosphatidylinositol-4-OH kinase (PI4K)-specific inhibitor KDU691 which kills dormant rings [181] or the imidazolopiperazines which kill both rings and dormant rings of artemisinin resistant parasites[182].Of note,the imidazolopiperazine KAF156 showed clinical efficacy in patients infected with parasites harbouring mutations inPfK13 propeller domain [137]. Further, it may be beneficial to think laterally and examine all possible options. For instance, the highly promising plant-based artemisinin combination therapy(pACT)has not been exploited[183].Unlike man-made ACTs which tend to be mixtures of typically two different magic bullets against the parasite, the polypharmacies that medicinal plants provide unique combinatorial therapies that are constituted by just one magic bullet combined with hundreds of other phytometabolites that often assist the magic bullet become a successful drug. The best example of this plant based combinatorial therapy is provided byArtemisnin annuaitself [4]. Thus, through an elegant set of experiments, it has been demonstrated that between an equal dose of artemisinin coming as pure drug vs as whole plant,the latter is significantly more potent than the former, significantly more bioavailable than the former when given through oral route, and three times less prone to develop artemisinin resistance than the former [183].
3.3. Long-lived gametocytocidal agents targeting stage V gametocytes should be prioritized
Stage V gametocytes are generally refractory to most antimalarial drugs [157]. This is the only stage of the parasite which can survive the mosquito mid-gut where fertilization occurs, and the parasite is thereafter transmitted [184].Although artemisinins can kill stage V gametocytes in vitro,they do not prevent transmission in vivo probably due to their short half-life [167,185-187]. Also,artemisinin-resistant parasites have a high propensity to form gametocytes [188]. This may explain the alarming spread of artemisinin resistance since it was first reported in 2008 [11,29,162].
Presently, our options for gametocytocidal agents with stage V gametocyte killing are limited to the 8-aminoquinolines which are toxic to individuals with glucose-6-phosphate dehydrogenase(G6PD) deficiency [189,190]. G6PD deficiency is prevalent in sub-Saharan Africa populations where it has been suggested to protect individuals from severe malaria[191]. However,the WHO has recommended a 0.25 mg/kg single dose of primaquine as the golden standard for transmission blocking which should not cause hemolysis in individuals with G6PD deficiency unlike the 14-day duration required for prevention of hypnozoite forms [10].
The inclusion of gametocytocidal agents will not only prevent the spread of resistance but could see us living in a malaria-free world. No one knows which of the antimalarials in development will first make it to clinical deployment.However,a good number of the clinical candidates in development are fast acting, have good safety profiles and have demonstrated stage V killing. Many of these drugs fit into more than one TCP of SERCaP.Combining them with complementary partners could contribute immensely to the eradication goal. With the level of recrudescence observed with ACT treatment [186,187], it suffices to say that a gametocytocidal agent with a long half-life may be extremely useful in curbing the transmission of parasites as it is transmission that plays a major a role in the rapid spread of resistance [11,162].
4. Concluding remarks
The fight against malaria cannot be won from within the confines of laboratories alone.Winning this battle will require all concerned parties to play their roles religiously. The recurring emergence of resistance to antimalarial drugs in the Greater Mekong sub-region suggests a need for stronger drug regulatory authorities that would ensure the enforcement of drug laws.Currently, there is a clamour for the elimination of malaria in this region before resistance renders the parasite immune to all the available antimalarials. On the one hand, the ban on monotherapies must be strictly enforced, and on the other hand,there must be strong legislation for prohibiting drug counterfeiting with stringent penal actions against perpetrators of drug crimes.
Secondly, it is apparent that strategies to monitor ACT failures must be different in low versus high transmission areas. While surveillance of K13 mutations is necessary in both low and high transmission regions, it appears that screening of molecular markers for partner drug resistance may be crucially important in high transmission areas as partner drugs may fail in these regions even before artemisinin resistance emerges. Hence, triple ACTs should be considered to delay the onset of resistance to dual ACTs and prevent the importation of resistance to these regions.
Lastly, even as our understanding of artemisinin resistance has improved tremendously in recent years, there appear to be more hidden players making it more complex than it already is. Certain gaps are still begging to be filled including determining whether all non-K13 mutations linked to artemisinin resistance are a result of mutations in K13-interacting proteins as well as understanding the characteristic phenotypes of non-K13 mutants to find whether they are different from what is known of K13 mutants. A better understanding of all players in artemisinin resistance is required for lasting strategies that will keep artemisinins therapeutically active until new drugs are ready to replace them as first-line agents.This may also invite better ideas to protect future drugs from resistance before they eventually get deployed.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
Nekpen Erhunse is supported by ICGEB Arturo Falaschi Predoctoral fellowship.
We thank the anonymous reviewers for their probing questions and critical comments that helped us elevate the key messages of this article.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2020.07.005.
 Journal of Pharmaceutical Analysis2021年5期
Journal of Pharmaceutical Analysis2021年5期
- Journal of Pharmaceutical Analysis的其它文章
- A sensitive electrochemical detection of metronidazole in synthetic serum and urine samples using low-cost screen-printed electrodes modified with reduced graphene oxide and C60
- Capsid destabilization and epitope alterations of human papillomavirus 18 in the presence of thimerosal
- Transformation of berberine to its demethylated metabolites by the CYP51 enzyme in the gut microbiota
- Acid-base and lipophilic properties of peptide nucleic acid derivatives
- Simulation of the oxidative metabolization pattern of netupitant, an NK1 receptor antagonist, by electrochemistry coupled to mass spectrometry
- One extraction tool for in vitro-in vivo extrapolation? SPME-based metabolomics of in vitro 2D,3D,and in vivo mouse melanoma models