
基于异质分类器集成的蛋白质结晶倾向性预测
2021-11-11 06:08:34梁亮
南京理工大学学报 2021年5期
梁 亮
(四川师范大学 网络与通信技术研究所,四川 成都 610066)
蛋白质的功能与其结构之间存在着密切的关系[1]。因此,蛋白质结构的准确解析对于理解生命活动的过程具有重要意义[2-4]。
X射线晶体衍射技术(X-ray crystallography)是解析蛋白质三维结构最主要的手段之一。截至2021年4月28日,蛋白质数据库(Protein data bank,PDB)中共计177 219个蛋白质结构,其中156 097个结构是利用X射线晶体衍射技术解析的,约占总数的88.1%。然而,在实际的蛋白质结构解析过程中,X射线晶体衍射技术的成功率只有10%左右[5]。其主要原因是大量实验蛋白质无法得到可供衍射的晶体,导致大量的时间和资源浪费在那些无法结晶的蛋白质上。另一方面,随着现代测序技术的飞速发展,蛋白质序列快速累积,结构和序列之间的鸿沟日益增大[5,6]。鉴于此,若能直接从蛋白质序列出发来准确预测其结晶倾向性,则对于提高基于X射线晶体衍射技术测定蛋白结构的成功率以及缩小蛋白质序列与结构之间的鸿沟将有重要价值。
已经有不少研究人员展开了从蛋白质序列出发预测接近倾向性的研究[7]。例如,Overton和Barton[8]根据等电位点(Isoelectric point,pI)和疏水性特征,开发了一个标准化的靶位分级量表,称为OB评分,可以用于结晶倾向性的评估。研究还发现,蛋白质的一些特定二级结构,如α螺旋、无规则卷曲等对蛋白质的结晶有影响[9-11]。随后,机器学习方法开始逐步被应用于蛋白质结晶倾向性的预测:例如Smialowski等[12]提出了一种SECRET方法,该方法基于蛋白质序列特征,综合使用支持向量机和朴素贝叶斯分类器来进行蛋白质结晶倾向性的预测;Kurgan团队也发表了一系列基于机器学习的蛋白质结晶倾向性方法,如CRYSTALP[13]、PCCpred[14]、MetaCrys[8]、CRYSTALP2[15]以及CRYSpred[16];此外,其他比较流行的基于机器学习的方法还包括:PXS[17]、XtalPred[18]、MetaPPCP[8]、ParCrys[19]、SVMCRYS[20]、PPCinter[21]、RFCRYS[6]、CMCRYS[22]以及TargetCrys[7]等等。
调研文献可以发现,从序列出发预测蛋白质的结晶倾向性已经取得了不小的进展,但是在精度上仍有提升的空间。本文提出了一种基于异质分类器集成的蛋白质结晶倾向性预测方法。该方法从蛋白质的组成成分以及进化信息视角抽取不同的特征并进行组合;然后,基于所抽取的特征训练多个异质分类模型;最后,将多个异质分类模型的输出进行集成作为最终的输出。在公开数据集上的评测结果表明,所提的蛋白质结晶倾向性预测方法是有效的,是对现有方法的一种有效补充。
1 特征抽取
要使用机器学习技术来进行蛋白质结晶倾向性的预测,首先要做的工作是将蛋白质序列进行特征表示[23,24]。特征表示的优劣对于后续分类器的性能有着至关重要的影响。本文将从蛋白质序列抽取4种典型的特征用于蛋白质结晶倾向性的预测。
1.1 氨基酸组成成分特征
用A1,A2,…,A19,及A20表示20种基本氨基酸。对于一条给定的长度为L的蛋白质序列,ci(ci,1≤i≤20)为氨基酸Ai在蛋白质序列中出现的次数。那么,该序列的氨基酸组成成分(Amino acid composition,AAC)特征是如式(1)所示的一个20维特征向量
(1)
1.2 伪氨基酸组成成分特征
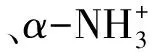
1.3 伪位置特异性得分矩阵特征
蛋白质的位置特异性得分矩阵(Position-specific scoring matrix,PSSM)编码了其进化信息,该信息可用于蛋白质属性(如结晶倾向性)的预测。对于一条给定的长度为L的蛋白质序列,使用PSI-BLAST[27]搜索Swiss-Prot数据库(E-value取值为0.001)可以得到其PSSM,为一个L×20的数值矩阵,记为:SPSSM=(si,j)L×20。
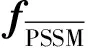
(2)
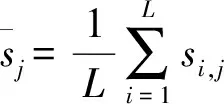
和AAC特征一样,上述PSSM组成成分特征同样丢失了序列中残基之间的顺序信息。因此,使用和PseAAC类似的方法,在原始PSSM的每一列上计算每个残基得分之间的相关性,从而得到伪位置特异性得分矩阵特征PsePSSM,过程如下[28,29]:
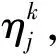
(3)
至此,PSSM的k阶相关性特征可以用式(4)表示
如今已是西藏大学理学院教授、钟扬的学生拉琼发现,病后稍有恢复的钟老师开始变本加厉地工作,一天排满了各种事。他的衣袋里总是装着很多小纸片,上面密密麻麻写满各种待办事项,每做完一项就用笔划掉。
(4)
最终的PsePSSM特征即式(5)所示
(5)
式中:K取值为6。因此,最终的PsePSSM特征维数为140(=20+6×20)维。
1.4 伪溶剂可及性特征
氨基酸残基的溶剂可及性(Solvent accessibility)是指蛋白质浸在溶液中时,溶剂分子与该残基的接触表面积。溶剂分子接触到的残基的面积越大,表明该残基暴露在蛋白质表面的部分越多;反之,则暴露的面积越小。已有的研究表明:蛋白质的微观表面性质对蛋白质的结晶行为有着至关重要的影响[30]。借鉴文献[31]的思想,本文亦使用伪溶剂可及性特征(Pseudo predicted solvent accessibility,PsePSA)来进行蛋白质结晶倾向性的预测。
对于一条给定的长度为L的蛋白质序列,首先使用工具软件SANN[32]预测出相应的溶剂可及性矩阵,记为PPSA=(pi,j)L×6。然后,采用和抽取PsePSSM特征一样的策略:
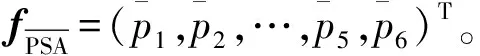
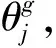
(6)
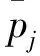
至此,PPSA的g阶相关性特征可以用式(7)表示
(7)
最终的PsePSA特征即如式(8)所示
(8)
式中:G取值为8。因此,最终的PsePSA特征维数为54(=6+6×8)维。
2 基于异质分类器集成的蛋白质结晶倾向性预测模型
图1给出了所提的基于异质分类器集成的蛋白质结晶倾向性预测模型框架。对于一条给定的待预测序列,分别提取相应的4种特征,亦即AAC、PseAAC、PsePSSM以及PsePSA;然后,将这4种特征进行串行组合,最终得到一个282维的特征向量;将此特征向量输入三个训练好的异质分类器,也就是支持向量机(Support vector machine,SVM)、随机森林[33](Random forests,RF)以及径向基神经网络(Radial basis function network,RBFN)。每个分类器均输出一个标量值,表示待预测蛋白质序列的结晶倾向性;随后,将这些分类器的输出进行集成。本文中,采用平均集成方案,即对每个分类器的输出进行平均后作为最终预测的蛋白质结晶倾向性。最后,使用一个阈值T*来判定待预测的蛋白质序列是否会结晶,亦即:如果预测的结晶倾向性大于阈值T*,则判定为可结晶;否则,判定为不可结晶。
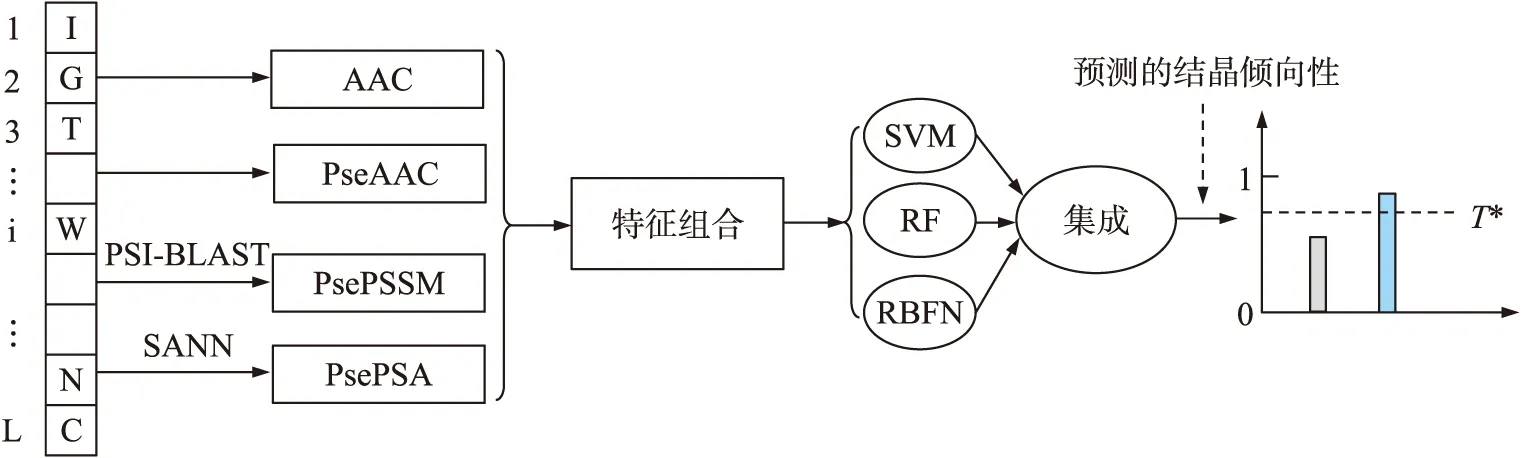
图1 基于异质分类器集成的蛋白质结晶倾向性预测框架
3 实验结果与分析
3.1 实验数据集及评价指标
为评估所述方法的有效性,本文采用文献[7]所使用的数据集。第一个数据集的训练集中包含756个正样本(可结晶蛋白质)和744个负样本(不可结晶蛋白质),记为TRAIN1500;其对应的独立测试集中包含244个正样本和256负样本,记为TEST500。第二个数据集的训练集中包含1 204个正样本和2 383个负样本,记为TRAIN3587;其对应的独立测试集中包含1 204个正样本和2 381个负样本,记为TEST3585。
使用敏感性(Sensitivity,Sen)、特异性(Specificity,Spe)、准确度(Accuracy,Acc)以及马修斯系数(Mathew’s Correlation Coefficient,MCC)这4个指标来度量预测模型的性能,具体定义如下
(9)
(10)
(11)
MCC=
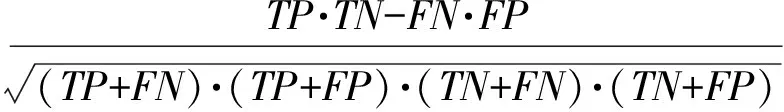
(12)
式中:TP、FN、TN以及FP分别表示测试结果中真阳性、假阴性、真阴性以及假阳性的样本数目。
3.2 结果与分析
为验证本文所提方法的有效性,在训练集上进行交叉验证并在独立测试集上进行泛化能力评估。
表1给出所提方法与其他3种基分类器方法(RBFN、SVM以及RF)在训练集TRAIN1500 和TRAIN3587 上五重交叉验证性能对比。
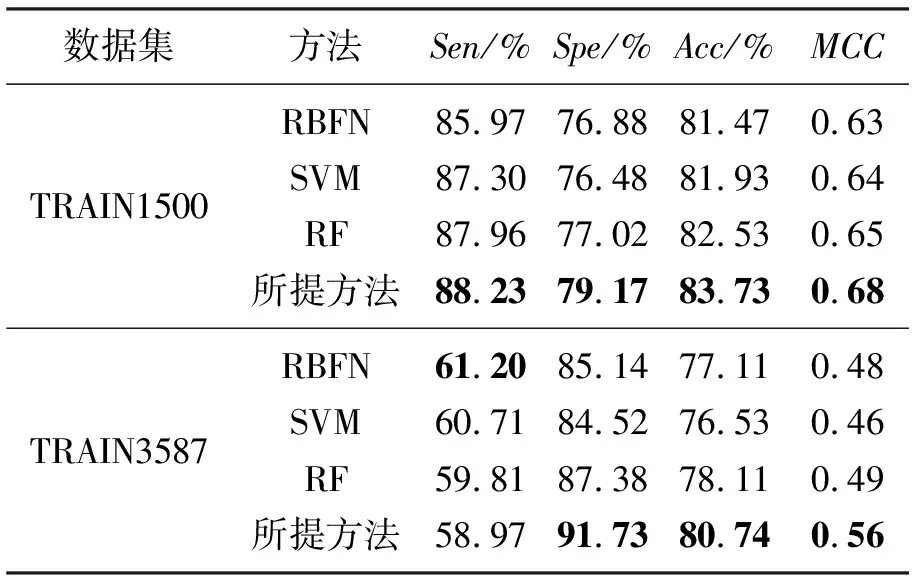
表1 所提方法与其他3种方法在训练集TRAIN1500和TRAIN3587上五重交叉验证性能对比
观察表1可以发现:在TRAIN1500数据集上,RBFN、SVM以及RF的MCC的分别为0.63、0.64以及0.65;在TRAIN3587数据集上,RBFN、SVM以及RF的MCC的分别为0.48、0.46以及0.49;其他3个指标,亦即Sen、Spe和Acc,3个方法在两个数据集上也取得非常接近的性能。这些结果表明:用于对比的3种方法在两个数据集上均取得了非常接近的预测性能。本文所提的方法在TRAIN1500和TRAIN3587的MCC值分别为0.68和0.56,比次优方法(RF)分别提高了4.6%和14.2%。在其他3个指标上,本文所提的方法也有不同程度的提升。实际上,本文所提的方法是对3种基分类器(RBFN、SVM以及RF)进行了集成,表1中的对比结果亦表明,集成后的方法比3种基分类器的性能均有了提高。这表明,3种基分类器的预测结果是具有互补性的。
为进一步验证本文所提方法的泛化能力,将其和3种基分类器方法在独立测试集TEST500和TEST3585进行了性能对比。需要注意的是,在测试TEST500时,本文方法和3种基分类器方法均是使用训练集TRAIN1500得到的;而在测试TEST3585时,所有的方法均是使用训练集TRAIN3587得到的。表2给出了所提方法与其他3种方法在独立测试集TEST500和TEST3585上的性能对比。观察表2,可以得到如下几个结论:首先,3种基分类器方法在两个独立测试集上同样得到了非常接近的结果;并且,对每种基分类器方法而言,在独立测试集上的结果和在对应训练集上的交叉验证结果非常接近。这表明对每个基分类器来说,其泛化能力没有被高估。其次,所提方法在两个独立测试上的性能一致性地高于3种基分类器方法。同样以MCC指标为例,所提方法在TEST500和TEST3585的结果分别为0.73和0.55,比次优方法(SVM)分别提高了8.9%和12.2%。
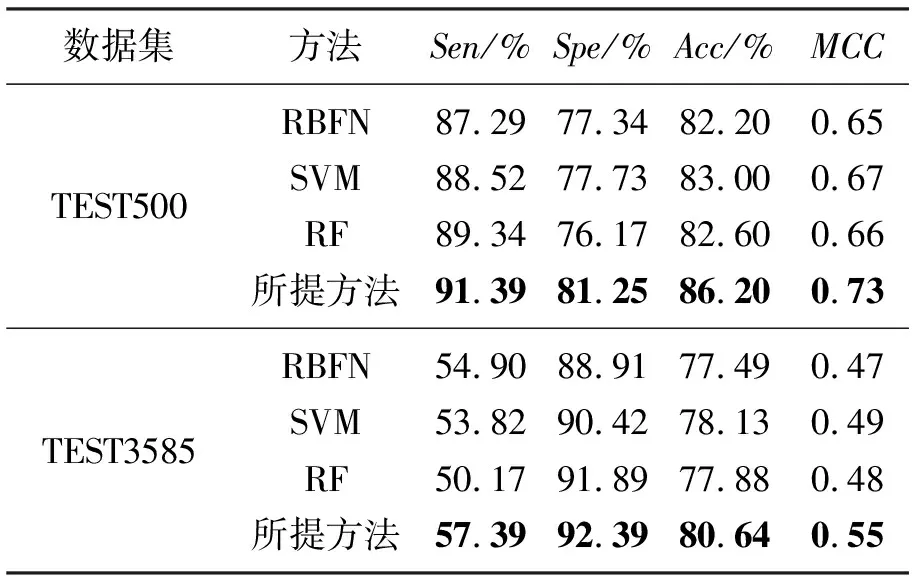
表2 所提方法与其他3种方法在独立测试集TEST500和TEST3585上的性能对比
综合表1及表2中的结果来看,通过对基分类器进行集成,所得到的集成模型对蛋白质结晶倾向性的预测能力进一步得到了提高。
表3给出了所提方法与其他9个用于蛋白质结晶倾向性预测的主流方法在独立测试集TEST3585上的性能对比,以进一步验证所提方法的有效性。由表3可以看出:首先,所提方法显著地优于前七种用于比较的方法,亦即MetaPPCP[8]、CRYSTALP2[15]、SVMCRYS[20]、XtalPred[18]、SCMCRYS[22]、PPCpred[14]以及RFCRYS[6];其次,所提方法的MCC值达到了0.55,和PPCinter[21]的MCC保持一致,略逊于用于比较方法中的最优方法TargetCrys[7]。这些结果表明,所提方法对蛋白质结晶倾向性的预测能力和最好的方法预测性能基本相当,也是对现有方法的有益补充。
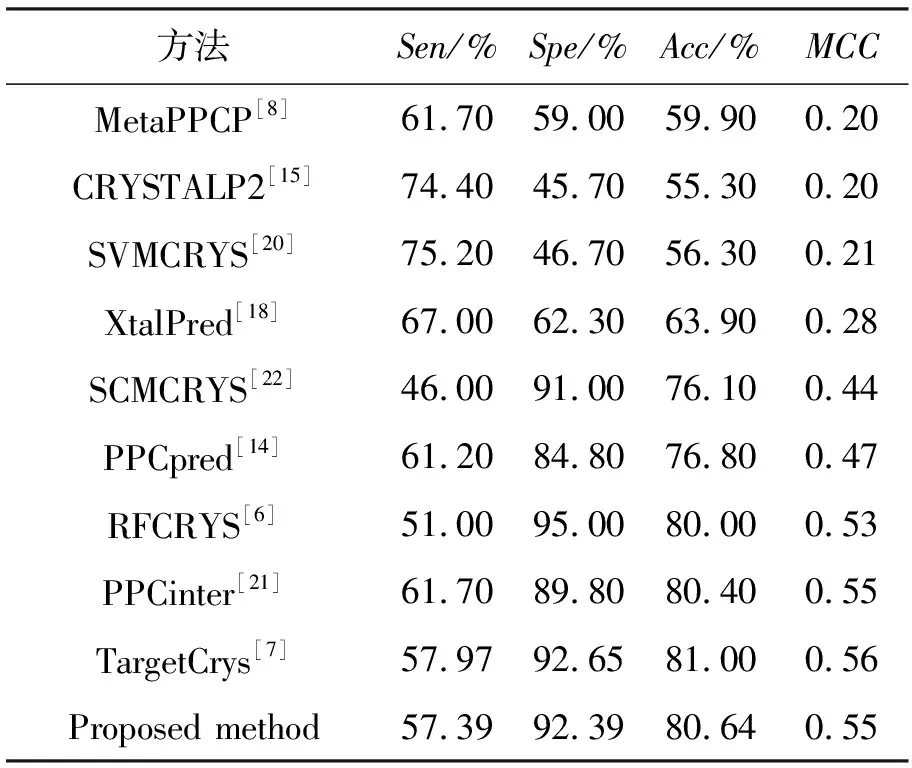
表3 所提方法与其他蛋白质结晶预测方法在独立测试集TEST3585上的性能对比
4 结束语
本文提出了一种基于异质分类器集成的蛋白质结晶倾向性预测方法。该方法从蛋白质序列出发,抽取多个视角的特征并进行组合;然后,基于所抽取的特征在训练集上训练多个不同的异质分类器并进行集成。在训练数据集上的交叉验证、独立测试以及和现有方法的对比结果验证了所提方法的有效性。在后续的工作中,拟从抽取更有鉴别力的特征、设计更为有效的分类模型以及提高模型的可解释性等方面展开研究,以进一步提升蛋白质结晶倾向性预测的性能。
猜你喜欢
有色金属(矿山部分)(2021年4期)2021-08-30 06:10:42
数学年刊A辑(中文版)(2020年2期)2020-07-25 02:04:44
数学物理学报(2019年6期)2020-01-13 06:08:16
数学物理学报(2017年5期)2017-11-23 07:51:31
云南师范大学学报(自然科学版)(2015年5期)2015-12-26 12:46:16
新闻研究导刊(2015年17期)2015-12-25 12:36:42
语言与翻译(2015年4期)2015-07-18 11:07:43
中央民族大学学报(自然科学版)(2015年2期)2015-06-09 08:45:26
物理实验(2015年10期)2015-02-28 17:36:52
中央民族大学学报(自然科学版)(2014年3期)2014-06-09 08:54:32
