
Ni、Co、Al掺杂尖晶石LiMn2O4的第一性原理计算
2021-11-10 02:25李旭王建川杜勇
粉末冶金材料科学与工程 2021年5期
李旭,王建川,杜勇
Ni、Co、Al掺杂尖晶石LiMn2O4的第一性原理计算
李旭,王建川,杜勇
(中南大学 粉末冶金国家重点实验室,长沙 410083)
针对分别掺杂Ni、Co、Al的尖晶石LiMn2O4,采用基于密度泛函理论的第一性原理计算方法,分析反铁磁层和铁磁层沿[001]方向交替排列的磁性构型的合理性。结果表明,此磁结构下表现出Mn3+/Mn4+沿[001]方向交替排列的价态分布。Ni、Co、Al三种掺杂原子都倾向于替换Mn3+层上的Mn,占据16d位后价态表现为Ni2+、Co3+和Al3+。Al与Ni掺杂均能抑制掺杂点位的Jahn-Teller畸变,且Ni掺杂后其最近邻的Mn3+被氧化为Mn4+,使结构更稳定,而Co掺杂可能导致更严重的Jahn-Teller畸变。Al3+和Co3+可显著降低Li离子的两条路径扩散能垒,Ni2+在Mn4+的协同作用下可进一步降低低能垒路径的能垒。
正极材料;LiMn2O4;掺杂;磁性结构;扩散
尖晶石结构的LiMn2O4(LMO)是一种典型的锰基正极材料[1-2],具有高电压平台(>4 V)以及较高的理论容量密度(148 mAh/g)和较好的热安全性,更重要的是锰具有成本低、无毒等优势。LMO的立方尖晶石结构属于Fd3m空间群,O原子位于32e位,呈立方体密集排列,Li离子位于8a四面体位,Mn离子占据16d八面体位的一半,另一半的八面体间隙为16c点位,是锂离子迁移的三维通道[3]。LMO中Mn的平均价态为+3.5,Mn3+和Mn4+等比例混合,其充放电活性来源于Mn3+/Mn4+的氧化还原[4]。在充放电过程中,Mn3+引起Jahn-Teller (JT)效应[5-6],导致其结构扭曲和相变等。此外Mn3+还产生歧化反应(2Mn3+→Mn4++ Mn2+),而Mn2+在电解液中容易溶解,导致容量严重衰减。
掺杂作为材料改性的一种重要方法,已广泛应用于锂离子电池电极材料的性能调控。在尖晶石LMO中掺杂可有效减少Mn3+引起的Jahn-Teller效应,而增加有利于结构稳定的Mn4+并抑制Mn2+的溶解,是改善充放电循环性能的关键。一般化学元素掺杂常用的元素有Al、Ni、Co、Cr、Zn和Mg等[7-11],掺杂元素的作用跟掺杂导致尖晶石结构的微小变化和晶体中元素价态变化有关。Ni-Co-Mn和Ni-Co-Al作为两类常见的三元材料,在层状结构下具有很好的协同作用,且将三种元素分别小计量单一掺杂进尖晶石结构的LMO中时也能起到很好的效果。如LEE等[11]、LIU等[7]、LI[8]和ARORA[9]等分别在LMO中掺杂少量Al、Ni和Co,均获得具有良好电化学性能和常温循环性能的LMO材料。这些掺杂都是掺杂入16d晶格位 置,可能会提升LiMn2O4的某种性能,但同时牺牲其它性能,如导致初始容量下降等。并且实验过程耗时较长、成本较高,掺杂方式也从单一的阳离子、阴离子掺杂演变为双离子、多离子复合掺杂,而且均缺乏对机理的定量解释。随着计算技术的发展,计算模拟成为研究掺杂的有效方法[12-14]。IDEMOT等[12]利用第一原理计算研究了LiNiMn2-xO4的结构性质,发现Ni掺杂使LiMn2O4的O-2p轨道和Mn-3d轨道的重叠增加,能形成更强的共价键,从而使晶体结构更加稳定。LEE等[13]将第一性原理计算与团簇展开方法相结合,建立了解释LiNi0.5Mn1.5O4中复杂阳离子排序的模型,可解释有序(P4332)和无序(Fd3m)Ni/Mn构型中间基态的差异。目前关于尖晶石LiMn2O4的掺杂性质的计算主要集中在掺杂对晶体结构和电子结构的影响方面,缺乏对Li离子在掺杂体系中扩散的认识,而锂离子扩散是电池性能的一个重要参数。此外,不同的磁性结构会影响LMO的基态结构及电荷分布,所以在材料研究中选择合理的磁性结构尤为关键,是获得准确结论的前提。而实际计算中出现大量磁性结构不统一的现象,LEE等[10]采用的是沿(110)方向的AFM (antiferromagnetic)磁性构型,KOYAMA等[15]和HUANG等[16]只是假设Mn的一个简单的高自旋FM(ferromagnetic)构型来计算和研究尖晶石LMO中的点缺陷。LIU等[17]对不同结构和磁性的LMO进行了更广泛的计算以找到其基态,使用8种不同的AFM和2个FM结构,包括OUYANG[18]使用的3个结构,他们发现最稳定的结构是AFM层和FM层沿[001]方向交替排列的高度对称构型,如图1(a)所示。因此需要在综合考虑其磁性结构的合理性基础上进行掺杂研究。基于以上分析,本文作者采用基于密度泛函理论(density functional theory, DFT)的第一性原理计算方法,研究LMO中AFM层和FM层沿[001]方向交替排列的磁性构型的合理性,在此基础上研究Al、Ni、Co三种元素微量掺杂尖晶石LMO后在体系内的状态,以及掺杂元素对LMO的晶体结构和电子结构的影响,最后用Cl-NEB方法讨论掺杂对锂离子扩散的影响,旨在从原子尺度探究LMN的掺杂性质,为LMO的掺杂研究提供理论指导。
1 计算方法
本文的所有DFT计算都在从头计算软件包VASP (vienna ab-initio simulation package)中完成[19-20]。价电子与核电子的相互作用采用投影缀加平面波方法(projector-augmented wave method,PAW)描述,电子间的相互作用采用Perdew-Wang的广义梯度近似(general gradient approximation, GGA)并通过Hubbard参数修正(即GGA + U)[21-23]。根据文献[24-26],Ni、Co、Mn的d轨道的U值分别设置为6.0、3.3和3.9 eV。Li、Mn、O、Ni、Co和Al等的价电子构型分别为1s22s1、3p63d54s2、2s22p4、3p63d84s2、3d74s2和3s23p1,其余电子均作为芯电子。为了将周期性边界条件对缺陷的影响降至最低,计算采用含有56个原子的Li8Mn16O32超胞,平面波的能量截止设定为500 eV,以保证总能收敛到1 meV/atom。对布里渊区的积分采用Monkhorst-Pack方案进行特殊的k点采样。对于结构驰豫,采用2×2×2的取样方案,当作用在每个原子上的剩余力小于0.2 eV/nm时达到优化的精度。总能量和电子结构的计算采用4×4×4的k点采样。考虑到掺杂缺陷在晶胞中的浓度较低,在掺杂晶胞弛豫过程中,将晶胞的体积和形状固定为无缺陷优化后的状态,只有内部原子的坐标完全弛豫。采用CI-NEB方法[27-28]进行离子扩散的计算。
2 结果与讨论
2.1 晶体结构与磁结构
根据LIU等[7]的预测,LiMn2O4的基态磁结构为沿[001]方向的AFM(↑↓↑↓)层和FM层交替的高度对称结构。在自旋极化条件下完全驰豫LMO结构,得到其基态原子结构和磁结构,如图1(a)所示,其中Mn的磁矩方向由箭头表述。经过结构优化后,立方晶胞沿方向拉长,变形为四方晶胞,晶格伸长率为7%,具体的晶格参数为==0.820 nm,=0.874 nm,与前人的实验数据(==0.811 nm,=0.865 nm)[16]和计算结果(==0.820 nm,=0.876 nm[17]或==0.822 nm,=0.876 nm[29])一致。因此尖晶石LMO是正交晶格而不是立方晶格。磁性结构中沿[001]方向在胞内存在2种不同的Mn-O6八面体结构,这2种八面体结构和Mn—O键长如图1(b)所示。Mn4+的Mn-O6八面体结构的Mn—O键长在/平面为0.193 nm,在方向为0.196 nm,近似为正八面体。而Mn3+的Mn-O6八面体整体有较大的畸变,/平面上的4个Mn—O键长基本相等(0.196~0.197 nm),而方向上的2个Mn—O键长相对于/平面的Mn—O更长(分别为0.223 nm和0.218 nm)。如前所述,Mn3+离子会引起JT变形,即沿方向有一定程度的拉伸,因此可以判断图1(a)中最上面的黄色Mn层为Mn3+层,其下面的紫色Mn层为无明显畸变的Mn4+层。JT畸变的Mn3+离子的Mn-O6八面体的Mn—O键长与300 K时的实验数据吻合较好(分别为0.192和0.226 nm)[30]。经计算,Mn3+离子的磁矩为35.43×10-24A∙m2,Mn4+离子的磁矩约为29.03×10-24A∙m2,这也与文献报道的高自旋Mn3+的磁矩为36.17×10-24A∙m2、低自旋Mn4+的磁矩为29.60×10-24A∙m2吻合较好[17]。磁矩的分析可进一步证实图1(a)中黄色Mn层为Mn3+层,紫色Mn层为Mn4+层。从图1(b)中Mn离子3d轨道上的电子分布示意图可知,引起Mn-O6八面体结构畸变的Mn3+是高自旋离子,而Mn4+是低自旋离子(两者实际都为高自旋状态,此时的高与低为相对而言),因此图1中黄色的Mn确实处于+3价状态。
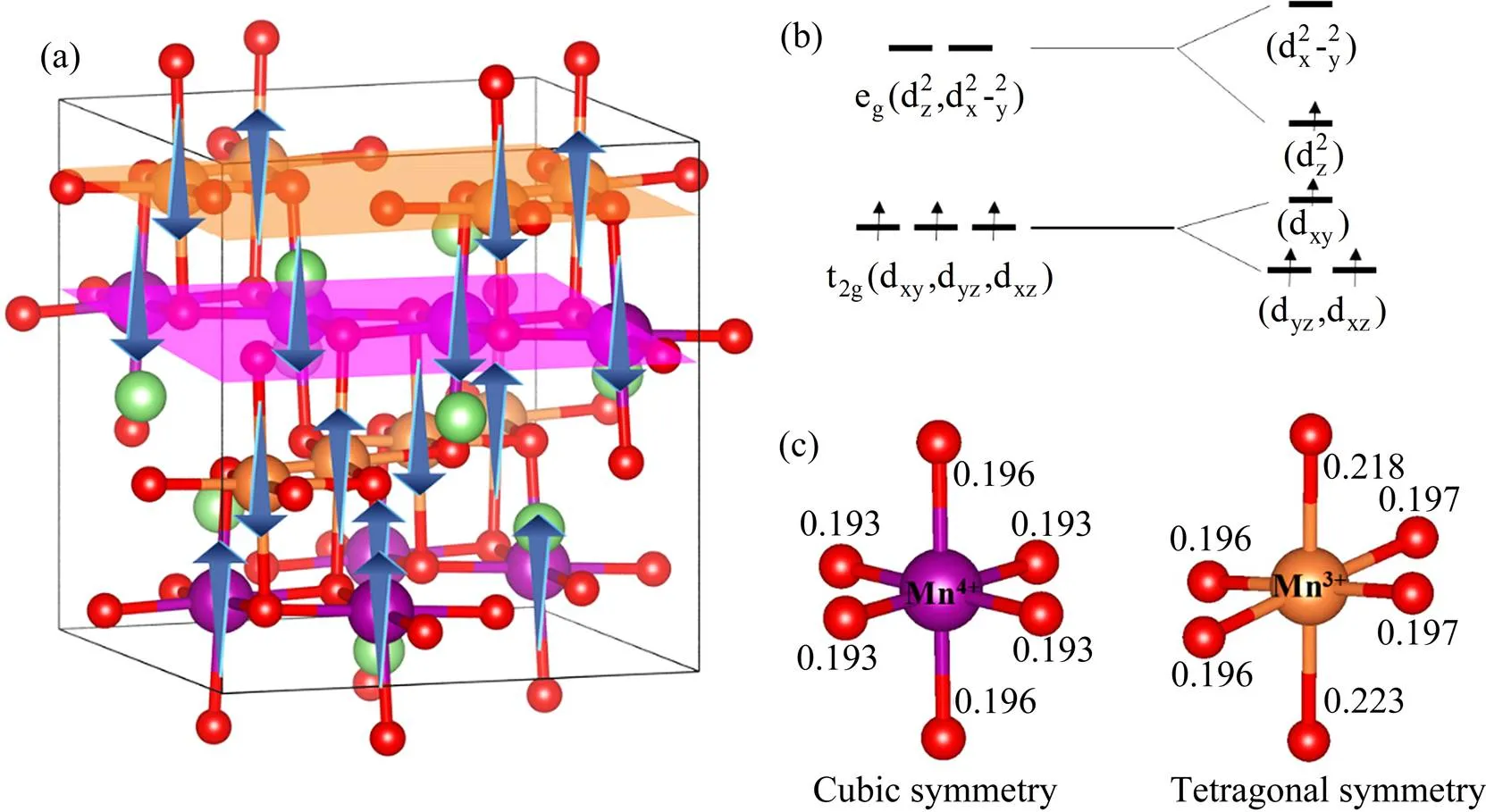
图1 尖晶石型LiMn2O4的晶体结构以及Mn3+/Mn4+离子的3d轨道电子分布与Mn3+-O6、Mn4+-O6八面体的结构参数
(a) The atomic structure of LiMn2O4and the magnetic configuration of Mn (the green and red spheres are Li and O, and the yellow and purple spheres are Mn3+and Mn4+, respectively. Arrow indicates the direction of magnetic moment of Mn); (b) Schematic diagram of electron distribution of 3d orbital of Mn3+and Mn4+ions; (c) Parameters of Mn3+-O6and Mn4+-O6octahedrons

2.2 掺杂形成能及掺杂体系的电子结构
考虑掺杂原子Ni、Co和Al取代LiMn2O4中16 d点位的Mn,掺杂缺陷形成能的计算公式为:
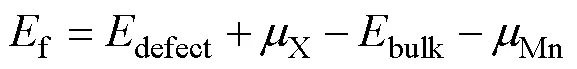
式中:Ef即掺杂缺陷形成能;Ebulk和Edefect分别为完美晶胞和含掺杂的缺陷晶胞的总能量;μ为单个原子的化学势。掺杂缺陷的产生涉及向晶胞中替换原子,式(1)中的μMn为Mn原子的化学势,μX为掺杂原子X的化学势。原子的化学势与该原子所处的平衡环境有关。本研究定义Mn、Co、Ni、Al的化学势为标准状态下处于稳定的单质状态时平均一个原子的能量。Al和Ni的基态构型均为FCC,Co的基态构型为HCP;对于Mn的化学势,本文作者在SLIWKO[31]的理论和实验工作的基础上采用I43 m结构进行计算,Mn占据3个不同的Wyckoff位置,并且每个位置的磁矩不同。LiMn2O4结构中Mn3+与Mn4+交替排列,所以掺杂原子X取代16d位的Mn原子存在两类情况,即取代Mn3+和Mn4+,分别记为和(上标III和IV分别表示X取代Mn3+和Mn4+)。表1所列为Al、Ni和Co在LiMn2O4结构中的掺杂形成能。从表中看出,掺杂原子取代Mn3+层上的Mn原子比取代Mn4+层上的Mn原子具有更低的形成能,也就是说这些掺杂原子倾向于取代+3价的Mn。值得注意的是,Al的掺杂形成能为负值(-1.4 eV),表明Al掺杂可自发的完成而不需要额外的能量。而Ni和Co掺杂需要额外能量,这些能量由掺杂实验中提供。
(a) The charge density of states for Mn4+ion’s 3d orbital; (b) The charge density of states for Mn3+ion’s 3d orbital; (c) The charge density of states for O ion’s 2p orbital
Note: The dotted and solid lines in the bottom figure represent the Px+Pyand Pzorbitals of O and the red and blue line indicate the O in Mn3+and Mn4+environment, respectively

表1 LiMn2O4中Al, Ni和Co的掺杂形成能
图3(a)所示为掺杂元素Ni、Co、Al分别与O构成的八面体结构。从图中看出,Al—O键长比掺杂前Mn3+—O的三个方向上的键长均短,且Al—O的方向键长(0.200 nm)比/方向键长(0.191~0.192nm)拉伸约4.2%。相比于纯LMO中11.7%的JT畸变,Al掺杂后JT畸变有较大的缓解,且Al—O键长较短。Co掺杂对原本的Mn3+-O6八面体结构影响不大,掺杂后/方向和方向的Co—O键长分别为0.193~0.194 nm和0.215~0.219 nm,方向畸变为11.6%,掺杂未表现出结构上的稳定作用。Ni掺杂也减弱了JT效应,Ni-O6八面体结构在/方向的键长为0.204~0.206 nm,比Mn3+—O键长有所增大,而在方向的键长小幅缩短,为0.214 nm和0.211 nm,其增加率约为3.6%。更重要的是,Ni掺杂直接影响Ni最近邻的一个Mn3+离子的状态,使得该Mn3+离子的磁矩由35.43×10-24A∙m2减小至29.03×10-24A∙m2,6条Mn—O键长在0.191~0.199 nm之间,该Mn3+离子的键长与磁矩近似于Mn4+。由此判断掺杂点位的Ni得到来自附近Mn3+的电子,表现为二价Ni状态,而Al和Co掺杂则没有此类现象,均为三价状态。

图3 掺杂原子晶体结构和对应的3d轨道电荷态密度图
(a) Crystal structures of Ni-O6, Co-O6and Al-O6octahedrons; (b), (c) 3d orbital’s charge density of state diagrams of doped Co and Ni atom respectively
图4所示为LMO总的态密度分布图和掺杂后的Mn-3d和O-2p轨道态密度图。从图4(a)发现锂离子Li-1s和O-2s轨道位于能级深处,受环境影响小,Li离子表现出强烈的离子性,掺杂后的电荷态密度分析将着重于费米能级附近。通过对比图4(b)、(c)和(d)发现,掺杂Ni和掺杂Al后总的电荷态密度变化都非常小,表现出在较低的掺杂浓度下,掺杂原子Ni和Al对整个体系的电荷态密度影响有限。但Co掺杂使得费米能级上方O-2p和Mn-3d的轨道向下移动0.5 eV左右,结合其在费米能级下方-0.5 eV处态密度峰升高,发现少量Co掺杂并不能缓解体系的JT效应,反而可能加重eg轨道的分裂,导致能量下降。
2.3 Li离子扩散
Li离子的扩散是电极材料充放电过程中的一个重要环节。对于尖晶石结构LMO,Li离子的扩散由空位机制引导,即Li离子通过8a→16c→8a这条三维扩散通道在两个8a位点之间完成一系列迁跃。图5所 示为Li离子的扩散通道和扩散能垒图。从图5(a)可知,Li离子在相邻的两个8a格点迁移需通过一个由6个Mn3+/Mn4+离子组成的“环”,这个“环”的中心就是16c点位。通常情况下由6个Mn3+/Mn4+离子组成的Mn6环有2种,如图5(a)所示,将离子通过由4个Mn4+和2个Mn3+组成的Mn6环的扩散路径命名为路径1,从[001]方向看,16c位点的中心位于Mn3+层上。Li离子通过由2个Mn4+和4个Mn3+组成的Mn6环的迁移路线称为路径2。从[001]方向看,路径2的16c位点位于Mn4+层上。本文采用CI-NEB方法,通过插入5个扩散中间态构型来研究Li离子在2个不同扩散路径上的扩散行为。
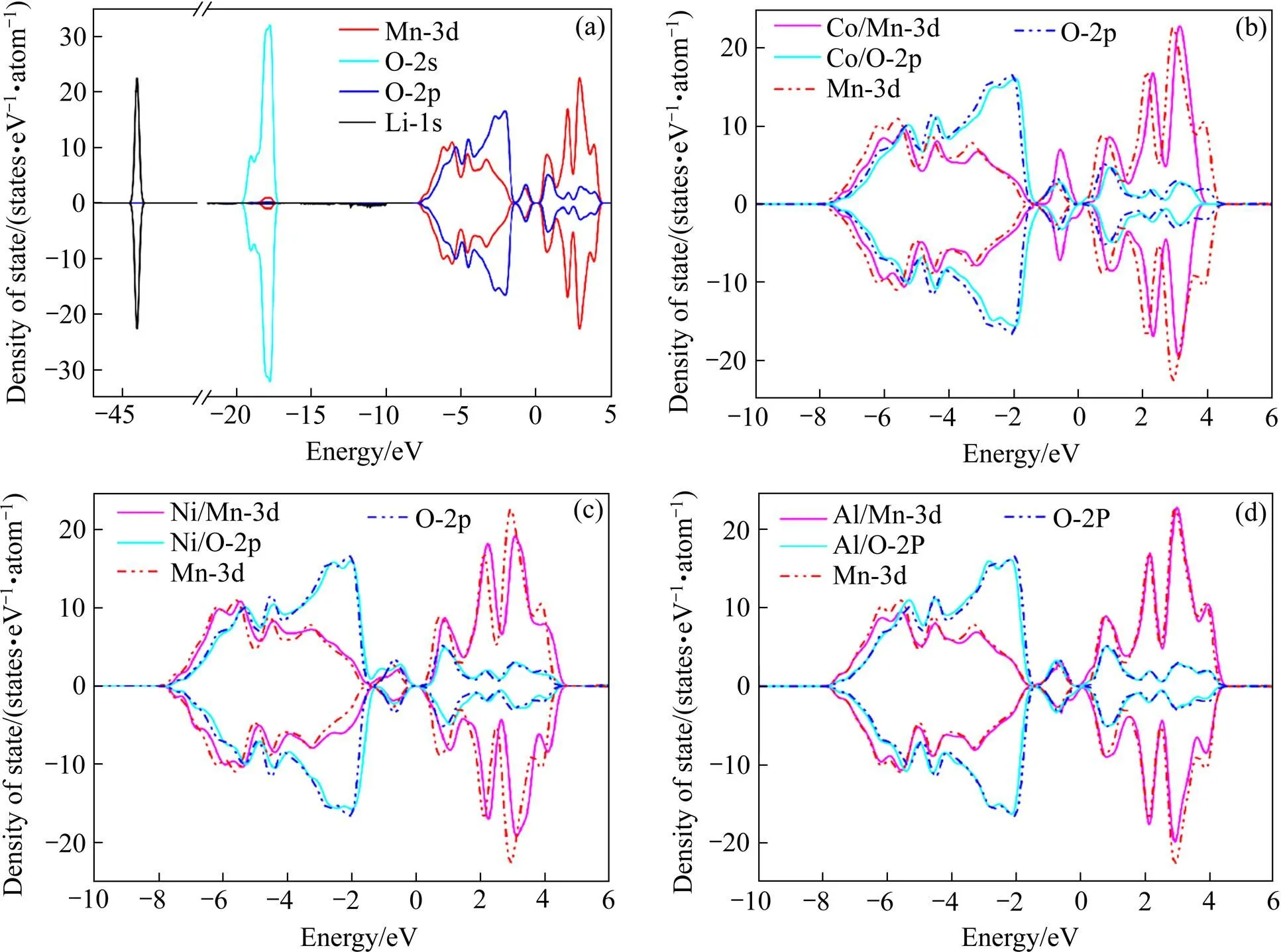
图4 LMO总的态密度分布图和掺杂后的Mn-3d和O-2p轨道的电荷态密度(实线)和未掺杂电荷态密度(虚线)对比图
(a) The total density of states of LMO (in (b), (c) and (d) change into the dotted line for comparison); (b), (c), (d) Comparison of charge density of state of Co, Ni , Al doped structures and undoped structures for Mn-3d and O-2p orbital, respectively
图5(b)所示为Li离子在无缺陷的LiMn2O4中的扩散能垒。由于路径1和路径2中Mn的氧化态不同,导致16c八面体间隙的形状和大小不同,也使得迁移的Li与Mn6环上Mn的静电相互作用不同。从图5(b)清楚地看到Li离子在两条路径上的扩散能垒不同,在路径1和路径2的扩散能垒分别为0.35 eV和0.68 eV。本文基于相同的磁构型下计算的路径1和路径2的扩散能垒与前人的理论值基本一致[17, 29]。XIAO等[29]报道的2条路径的扩散能垒分别为0.38 eV和0.72 eV,LIU等[17]的研究的路径1和路径2的扩散能垒分别为0.23 eV和0.70 eV。此外当Li离子通过路径2扩散时,在Mn6环中心16c位置达到亚稳态构型,基态能量处于局部低点。对比两种路径的扩散能垒可知,Li离子更容易在Mn4+离子较多的路径1扩散,其扩散能垒较小。Li离子在路径2中的扩散能垒较高,归因于阳离子间的静电相互作用。与Mn3+相比,Mn4+的电子云范围较小,因此Mn4+离子对移动Li离子的库仑排斥作用较小,且Mn3+的畸变也造成扩散通道的结构收缩,因此,无缺陷的LiMn2O4中,在对称一致的情况下,Mn4+比例越高,Li离子的迁移能垒越低;引发JT畸变的Mn3+离子越多,扩散能垒越高。

图5 Li离子的扩散路径和扩散能垒图
(a) Two diffusion paths of LiMn2O4crystal structure; (b), (c), (d), (e) Diffusion energy barriers of Li ions along paths 1 and 2 in undoped LiMn2O4and in Co, Ni and Al doped LiMn2O4respectively

Co掺杂取代Mn3+点位后显示为三价,Co3+的离子半径为0.054 5 nm[33]。Co3+离子半径较小,故其对扩散Li离子的库伦相互作用较小,再加上Mn3+离子的JT效应减弱,所以Co掺杂后Li离子的两条路径的扩散能垒均显著降低,路径1和2 的扩散能垒分别为0.30 eV和0.59 eV。与未掺杂状态的1、2路径扩散能垒相比,分别下降0.09 eV和0.05 eV。Al既不是过渡金属,也没有磁性,从Al—O键长看出,Al-O6八面体基本未出现结构畸变。Al3+离子半径为0.053 5 nm[33],相比于Co3+和Mn3+的半径有所减小,所以Al掺杂后Li离子在路径1和2的扩散能垒相对于Co掺杂有小幅降低,分别为0.27 eV和0.56 eV。
综上所述,无论是无缺陷的LMO还是Al、Co及Ni掺杂的LMO,Li离子在路径1的扩散能垒都低于路径2,可将路径1视为Li离子的快速扩散通道。此外,Ni、Co、Al三种元素掺杂都可在一定程度上降低Li离子迁移的扩散能垒,其具体的影响效果与元素价态及离子半径相关。价态相同的Al3+与Co3+对离子扩散能垒的影响与离子半径相关,较小的离子半径能降低扩散能垒,而半径较大的Ni2+能氧化同层最近邻的Mn3+离子,从而减小Li离子通过路径1扩散时的扩散能垒,但对于路径2的扩散能垒无显著影响。
3 结论
1) 采用第一性原理计算方法,研究LMO中AFM层和FM层沿[001]方向交替排列的磁性构型的合理性,发现Mn3+离子和Mn4+离子沿[001]方向交替排列。在此基础上研究Ni、Al、Co三种元素掺杂尖晶石结构LiMn2O4的性质。
2) Ni、Co、Al三者都更倾向于取代Mn3+层上的Mn,占据16d点位后的掺杂原子分别表现为Ni2+、Co3+和Al3+价态。Al掺杂能够在掺杂点位缓解JT畸变;而Ni掺杂不仅能减弱掺杂点位的结构畸变,还能将其最近邻的Mn3+氧化为Mn4+,有利于结构的稳定;Co掺杂后依然有明显畸变。
3) Li离子的扩散能垒与扩散通道中掺杂元素离子半径及离子价态相关,Mn3+离子半径>Co3+离子半径>Al3+离子半径,离子半径越小,扩散能垒越低。Ni2+离子半径大,但其将同层最近邻的Mn3+离子氧化为Mn4+离子,在Mn4+离子的协同作用下,有效降低路径1的扩散能垒。
[1] GUMMOW R J, KOCK A D, THACKERAY M M. Improved capacity retention in rechargeable 4V lithium/lithium- manganese oxide (spinel) cells[J]. Solid State Ionics, 1994, 69(1): 59-67.
[2] GUYOMARD D, TARASCON J M. The carbon/Li1+xMn2O4system[J]. Solid State Ionics, 1994, 69(3/4): 222-237.
[3] TAKADA T, HAYAKAWA H, ENOKI H, et al. Structure and electrochemical characterization of Li1+xMn2-xO4spinels for rechargeable lithium batteries[J]. Journal of Power Sources, 1999, 81/82: 505-509.
[4] LIU T, DAI A, LU J, et al. Correlation between manganese dissolution and dynamic phase stability in spinel-based lithium-ion battery[J]. Nat Commun, 2019, 10(1): 4721.
[5] NAKAYAMA M, NOGAMI M. A first-principles study on phase transition induced by charge ordering of Mn3+/Mn4+in spinel LiMn2O4[J]. Solid State Communications, 2010, 150(29/30): 1329-1333.
[6] RODRIGUEZ-CARVAJAL J, ROUSSE G, MASQUELIER C, et al. Electronic crystallization in a lithium battery material: columnar ordering of electrons and holes in the spinel LiMn2O4[J]. Physical Review Letters, 1998, 81(21): 4660-4663.
[7] LIU G, WANG Y, LI W. Synthesis and electrochemical performance of LiNi0.5Mn1.5O4spinel compound[J]. Electrochimica Acta, 2005, 50(9): 1965-1968.
[8] GUOHUA L, IKUTA H, UCHIDA T, et al. The spinel phases LiMMn2-yO4(M=Co,Cr,Ni) as the cathode for rechargeable lithium batteries[J]. Journal of the Electrochemical Society, 1996, 143(1): 178-182.
[9] ARORA P, POPOV B, WHITE R E. Electrochemical investigations of cobalt-doped LiMn2O4as cathode material for lithium-ion batteries[J]. Journal of the Electrochemical Society, 1998, 145(3): 807.
[10] LEE Y K, PARK J, LU W. Electronic and bonding properties of LiMn2O4spinel with different surface orientations and doping elements and their effects on manganese dissolution[J]. Journal of the Electrochemical Society, 2016, 163(7): A1359-A1368.
[11] LEE Y S, KUMADA N, YOSHIO M. Synthesis and characterization of lithium aluminum-doped spinel (LiAlMn2-xO4) for lithium secondary battery[J]. Journal of Power Sources, 2001, 96(2): 376-384.
[12] ITO Y, IDEMOTO Y, TSUNODA Y, et al. Relation between crystal structures, electronic structures, and electrode performances of LiMn2-xMO4(M=Ni,Zn) as a cathode active material for 4V secondary Li batteries[J]. Journal of Power Sources, 2003, 119(8): 733-737.
[13] LEE E, PERSSON K A. Revealing the coupled cation interactions behind the electrochemical profile of LiNi0.5Mn1.5O4[J]. Energy & Environmental Science, 2012, 5(3): 6047-6051.
[14] BENEDEK R, JOHNSON C, THACKERAY M. First-principles calculations for Co-doped LiMn1. 5Ni0. 5O4and LiMn2O4battery electrodes[J]. Electrochemical and Solid State Letters, 2006, 9(6): A289-A291.
[15] KOYAMA Y, TANAKA I, ADACHI H, et al. First principles calculations of formation energies and electronic structures of defects in oxygen-deficient LiMn2O4[J]. Journal of The Electrochemical Society, 2003, 150(1): A63-A67.
[16] HUANG R, IKUHARA Y H, MIZOGUCHI T, et al. Oxygen- vacancy ordering at surfaces of lithium manganese(III,IV) oxide spinel nanoparticles[J]. Angewandte Chemie, 2011, 123(13): 3109-3113.
[17] LIU W W, WANG D, WANG Z, et al. Influence of magnetic ordering and Jahn–Teller distortion on the lithiation process of LiMn2O4[J]. Physical Chemistry Chemical Physics, 2017, 19(9): 6481.
[18] OUYANG C Y, SHI S Q, LEI M S. Jahn–Teller distortion and electronic structure of LiMn2O4[J]. Journal of Alloys and Compounds, 2009, 474(1/2): 370-374.
[19] A G K, B J F. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set-science direct[J]. Computational Materials Science, 1996, 6(1): 15-50.
[20] KRESSE G G, FURTHMÜLLER J J. Efficient iterative schemes for Ab initio total-energy calculations using a plane-wave basis set[J]. Physical Review B, 1996, 54: 11169-11186.
[21] PERDEW J P, YUE W. Accurate and simple analytic representation of the electron-gas correlation energy[J]. Physical Review B: Condensed Matter, 1992, 45(23): 13244-13249.
[22] WANG Y, PERDEW J P. Correlation hole of the spin-polarized electron gas, with exact small-wave-vector and high-density scaling[J]. Physical Review B: Condensed Matter, 1991, 44(24): 13298.
[23] ANISIMOV V I, ZAANEN J, ANDERSEN O K. Band theory and Mott insulators: Hubbard U instead of stoner I[J]. Physical Review B, 1991, 44(3): 943-954.
[24] KITCHAEV D, PENG H, LIU Y, et al. Energetics of MnO2polymorphs in density functional theory[J]. Physical Review B, 2016, 93(4): 045132-045138.
[25] SHIIBA H, ZETTSU N, NAKAYAMA M, et al. Defect formation energy in spinel LiNi0.5Mn1.5O4-δusing Ab initio DFT calculations[J]. Journal of Physical Chemistry C, 2015, 119(17): 9117-9124.
[26] JAIN A, ONG S P, HAUTIER G, et al. Commentary: The materials project: A materials genome approach to accelerating materials innovation[J]. APL Materials, 2013, 1(1): 011002 -011013.
[27] HENKELMAN G, JÓNSSON H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points[J]. The Journal of Chemical Physics, 2000, 113(22): 9978-9985.
[28] HENKELMAN G, UBERUAGA B P, JóNSSON H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths[J]. The Journal of Chemical Physics, 2000, 113(22): 9901-9904.
[29] XIAO W, XIN C, LI S, et al. Insight into fast Li diffusion in Li-excess spinel lithium manganese oxide[J]. Journal of Materials Chemistry A, 2018, 6(21): 9893-9898.
[30] YAMAGUCHI H, YAMADA A, UWE H. Jahn-Teller transition of LiMn2O4studied by X-ray-absorption spectroscopy[J]. Physical Review B, 1998, 58(1): 8-11.
[31] SLIWKO V, MOHN P, SCHWARZ K. The electronic and magnetic structures of alpha-and beta-manganese[J]. Journal of Physics Condensed Matter, 1994, 6(32): 6557-6564.
[32] MACNEIL D, LU Z, DAHN J R. Structure and electrochemistry of Li[NiCo1-2xMn]O2(0<<1/2)[J]. Journal of the Electrochemical Society, 2002, 149(10): A1332-A1336.
[33] SHANNON R D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides[J]. Acta Crystallographica Section A, 1976, 32(5): 751-767.
First-principles calculations of Ni, Co and Al doped spinel LiMn2O4
LI Xu, WANG Jianchuan, DU Yong
(State Key Laboratory of Powder Metallurgy, Central South University, Changsha 410083, China)
For spinel LiMn2O4doped with Ni, Co and Al, the first-principles calculations method based on density functional theory is used to analyze the magnetic configuration of the anti ferromagnetic layer and the ferromagnetic layer alternately arranged along the [001] direction rationality. The result show that this magnetic structure shows the valence distribution of Mn3+/Mn4+alternately arranged along the [001] direction. The doping calculation shows that all three kinds of atoms tend to replace Mn in the Mn3+layer, and the valence states of doped atoms are Ni2+, Co3+and Al3+after occupying the 16d site. Al and Ni doping can inhibit the Jahn-Teller distortion of doping sites, and its nearest neighbor Mn3+is oxidized to Mn4+after Ni doping, which is more conducive to the stability of the structure. Co doping may result in more severe Jahn-Teller aberrations. The Al3+and Co3+can significantly reduce the diffusion energy barrier of Li ion in two paths, and Ni2+further reduce the low energy barrier path’s energy barrier under the synergistic action of Mn4+.
cathode material; LiMn2O4; doping; magnetic structure; diffusion
O483
A
1673-0224(2021)05-387-09
中德合作研究小组项目(GZ1528)
2021-03-13;
2021-06-18
王建川,副教授,博士。电话:13080566873;E-mail: jcw728@126.com
(编辑 汤金芝)
猜你喜欢
山东冶金(2022年4期)2022-09-14
教学考试(高考化学)(2022年4期)2022-08-30
耐火材料(2022年4期)2022-08-28
华东理工大学学报(自然科学版)(2022年2期)2022-04-29
中国宝玉石(2022年2期)2022-04-25
陶瓷学报(2021年3期)2021-07-22
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
新世纪智能(数学备考)(2019年9期)2019-10-16
中国有色金属学报(2018年2期)2018-03-26
