
CDK1、CCNB1和NDC80与乙型肝炎相关肝细胞癌的预后和进展相关:基于生物信息学方法
2021-11-10 16:24李玉杰吴登强韦常宏杨雪佳周素芳
南方医科大学学报 2021年10期
李玉杰,吴登强,韦常宏,杨雪佳,周素芳
1广西医科大学基础医学院生物化学与分子生物学教研室,2长寿与老年相关疾病教育部重点实验室,3广西高校生物分子医学研究重点实验室,4区域性高发肿瘤早期防治研究教育部重点实验室,广西 南宁530021
肝癌是我国发病率第3位的恶性肿瘤,5年生存率仅为10%左右。据统计我国约有HBV 感染者1.2 亿,占我国总人口的9.09%,其中有1/4 是慢性乙肝患者[1,2]。目前可用的抗病毒药物不能完全消除慢性乙型肝炎病毒。慢性HBV感染可导致肝脏慢性炎症,导致正常肝细胞转化为癌细胞,使乙肝病毒成为重要的环境致癌物[3-5]。因此,为了提高患者的存活率,迫切需要有效的生物标志物。近十年来,随着全基因组基因表达芯片的广泛应用,基于基因表达谱检测出了多种分子标记,其中有几种已用于肝癌的临床治疗。这些标记物在早期诊断、分子分型、化疗敏感性和耐药性、预后预测和监测等方面具有重要价值[6,7]。这些技术是分析海量基因表达数据集的极佳选择,以便深入解释HCC的机制。目前,利用生物信息学方法可以识别与肿瘤消退相关的潜在生物标志物和信号通路。到目前为止,在大样本的基础上,还没有足够的生物信息学研究聚焦于HBV感染患者的HCC组织和非肿瘤组织之间的差异表达基因(DEGs),HBV促进HCC发生的确切分子机制还不完全清楚,相关生物标志物众多,无法为临床预后提供有针对性的依据[8]。迫切需要对相关的全基因组数据进行进一步的全面基因组分析[9,10]。因此,利用生物信息学分析,可以通过识别hub基因(与其他基因有大量相互作用,通常在信号通路和生物学过程中起重要作用的基因)来阐明HBV相关肝细胞癌的潜在分子机制,从而有助于开发有效的新的诊断和治疗策略。在本研究中,为了增加样本量,我们整合了从GEO数据库获得的3个数据集,使用一个大的队列,确定了HBV相关肝癌组织和非肿瘤组织之间的DEGs。并试图利用生物信息学分析来识别hub基因和通路,筛选HBV感染诱导的肝癌的潜在治疗靶点。
1 资料和方法
1.1 来自GEO的公开数据
基于GPL570 平台的3 个微阵列数据集([HGU133_Plus_2]Affymetrix Human Genome U133 Plus 2.0 Array)来 自GEO(http://www.ncbi.nlm.nih.gov/geo/)[11].GSE55092 数据集包含49个HBV相关的HCC组织样本和91个HBV感染样本,GSE121248数据集包含70个HBV相关的HCC组织样本和37个HBV感染样本,GSE84044数据集包含124个HBV感染样本。所有原始数据(CEL)均从GEO 数据库下载。使用Robust Multichip Average 调整背景的原始文件。探针集注解可在affymetrix 官网(http://www.affymetrix.com/support/technical/annotationfilesmain.affx)下 载,“sva”R包用于消除批间差。
1.2 筛选DEGs
使用基于未配对t检验的R 包“affy”(https://biocon-ductor.org/packages/affy/)鉴定HBV 相关HCC组织和非肿瘤HBV相关组织之间的DEG;阈值为|log2(fold change)|≥1.5,调整后的P<0.01。DEG由使用R包“gplots”(https://bioconductor.org/packages/gplots/)的热图表示。
1.3 DEGs的功能和通路富集分析
用于注释、可视化和集成发现的数据库(DAVID,https://DAVID.ncifcrf.gov/home.jsp)是基于GO(http://www.geneontology.org)和KEGG(https://www.kegg.jp/)[12-15]。DEG分为3大类:生物过程(BP)、细胞成分(CC)和分子功能(MF)。通路分析应用于KEGG通路富集。
1.4 PPI网络的构建与模块分析
检索相互作用基因/蛋白质的搜索工具(https://string-db.org/.11.0版)是一种用于识别DEG相互作用和功能关联的系统,构建蛋白质-蛋白质相互作用(PPI)网络。在Cytoscape(https://cytoscape.org/)上,CytoHubba插件用于获取在PPI 网络上得分最高的前10 个PPI hub 基因。
1.5 遗传标记的生存分析和临床特征
使用cBioPortal(http://www.cbioportal)分析关键基因之间的相关性。使用Oncomine 数据库(https://www.oncomine.org/)验证遗传标记的表达水平,该数据库是具有基于网络的数据挖掘平台的微阵列癌症数据库,以支持全基因组表达的分析。数据集按癌症类型(肝癌)和分析类型(癌症与正常)进行过滤。使用UALCAN 在线分析工具(http://ualcan.path.uab.edu/index.html)对遗传标记进行预后分析,该工具结合了来自TCGA 数据库的预后数据。Kaplan-Meier plotter(http://kmplot.com/analysis)是一个开源癌症大数据分析网站,提供生存生物标志物的在线验证并分析具有枢纽关键基因的患者的总生存期(OS)。
1.6 逆转录定量(RT-q)PCR
2018年5月~2020年9月在广西医科大学附属肿瘤医院进行的手术后,总共获得了17个HCC 组织和17个配对的相邻非肿瘤组织(表1)。所有组织在用于实验前均获得患者同意。本研究经广西医科大学伦理委员会批准,符合广西医科大学的伦理指南和规定。FFPE RNA分离试剂盒(Omega Bio-Tek,Inc.)。根据制造商的方案,使用PrimeScript RT-qPCR 试剂盒(Takara Bio,Inc.)获得互补(c)DNA。使用GoTaq qPCR Master Mix 和SYBR green I(Takara Bio,Inc.)进行PCR扩增。将mRNA 表达水平标准化为ACTB。细胞周期蛋白依赖性激酶1(CDK1)、细胞周期蛋白B1(CCNBI)和核分裂周期蛋白80(NDC80)的引物序列如下:CDK1 正向5'-AGC CGC CCT TTC CTC TTT CTT TC-3'和反向5'-CGG ATT CAC CAA TCG GGT AGC C-3';CCNB1正向5'-GCC AGT GCC AGA GCC AGA AC-3'和反向5'-CAT TGG GCT TGG AGA GGC AGT ATC-3';NDC80 正 向5'-GTG CCA GTG AGC TTG AGT CCT TG-3'和反向5'-CGT CTT TCT TCA GTC GTG GTT TGC-3';ACTB 正 向5'-AGG TCG GTG TGAACG GAT TTG-3'和反向5'-GGGGTCGTTG ATGGCAACA-3'(Sangon Biotech)。

表1 17例样本的基本临床信息Tab.1 Basic clinical information of 17 patients with HCC
PCR热循环条件如下:95 ℃初始变性30 s,95 ℃5 s和60 ℃34 s 40个循环。对于每个样品,在20µL反应体积中进行3 次重复实验,其中包含2 µL 稀释的cDNA、1.6 µL 正向和反向引物、10 µL SYBR Premix Ex Taq™(Takara Bio)和0.4µL ROX II Reference染料(Takara Bio)。CDK1、CCNB1 和NDC80 与ACTB 的相对表达使用2-ΔΔCt方法计算。
1.7 统计学方法
使用R-3.6.3和SPSS22.0(IBM Corp.)进行统计分析。计数资料比较采用χ2检验和Fisher's精确检验,连续资料比较采用t检验,绘制Kaplan-Meier曲线,并进行对数秩检验以评估患者生存。P<0.05被认为具有统计学意义。
2 结果
2.1 DEGs的分析
在3个数据集(GSE55092、GSE121248 和GSE84044)中,其中119个为HBV相关的HCC 组织,其中252个为HBV 相关的组织。整合了3个数据集进行分析并去除批间差效应(图1A)。总共确定了121个DEG,其倍数变化阈值>1.5 或<-1.5,调整后的P<0.01。图1B中提供了DEG的热图。
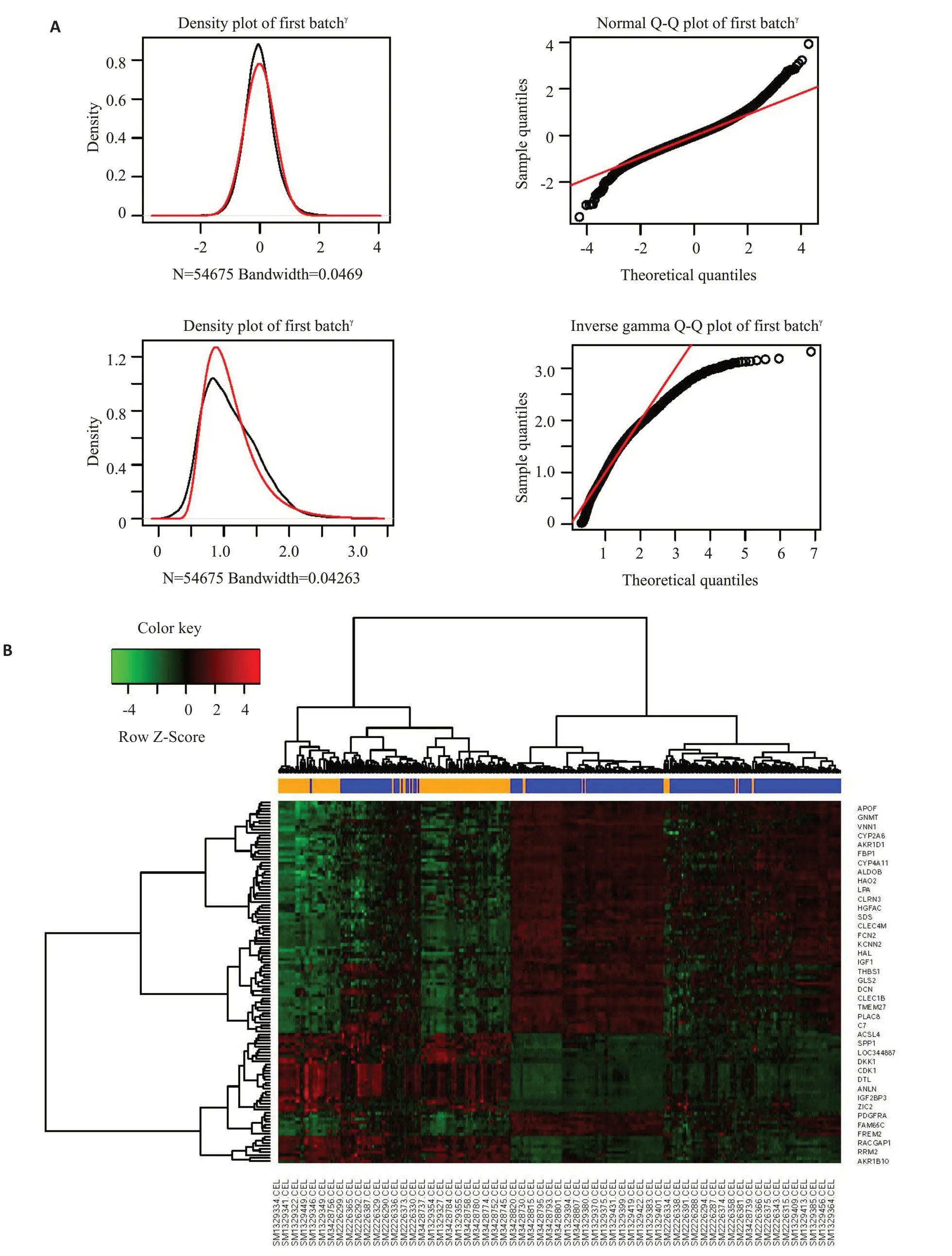
图1 去除微阵列数据中的批间差效应和DEG表达谱热图的构建Fig.1 Adjustment of batch effects in microarray expression data and heatmap of the expression profiles for the differentially expressed genes (DEGs).A:Removing the batch effects.B:Hierarchical clustering heatmaps of the DEGs screened on the basis of log|FC|>1.5 and a corrected P<0.01.The colors represent the expression level of the genes,and the higher the expression level,the darker the color(red,upregulated;green,downregulated).
2.2 DEGs的富集分析
记录了富集分析的前10个结果(图2)。BP 类别中显著富集的GO功能是氧化还原过程、外源药物分解代谢过程和环氧化酶P450 途径。MF类别中显著富集的GO功能是血红素结合、铁离子结合和氧化还原酶活性,作用于成对的供体,结合或还原分子氧。CC类别中显著富集的GO功能是细胞外区域、细胞器膜和中间体。对于KEGG通路,基因主要富集在视黄醇代谢、代谢通路和咖啡因代谢中。
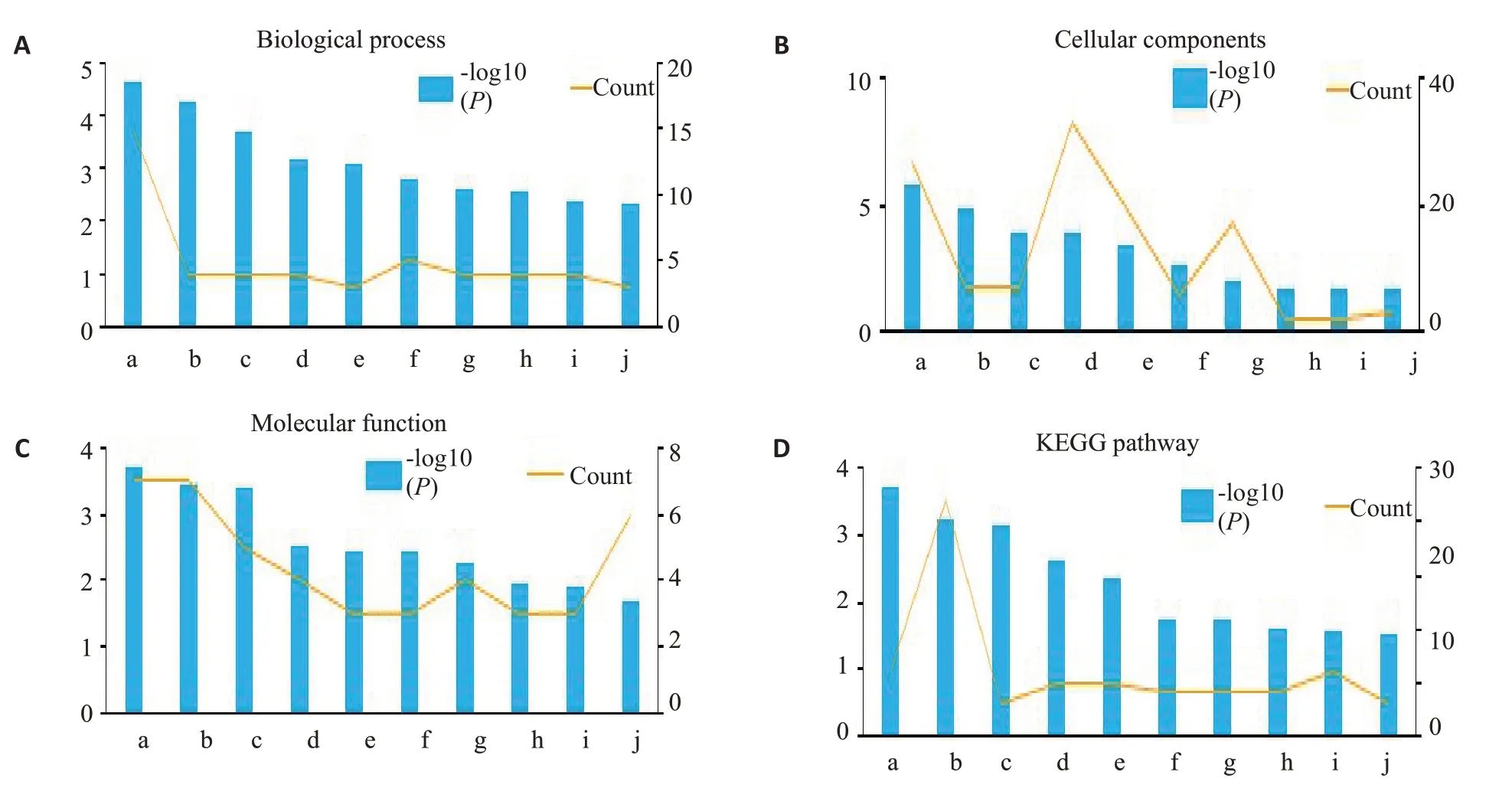
图2 DEG 的GO 和KEGG 通路富集分析Fig.2 GO and KEGG pathway enrichment analyses of the DEGs.A:Biological process (BP).B:Cellular component(CC).C:Molecular function (MF).D:KEGG pathway analysis.a:Oxidation-reduction process;b:Exogenous drug catabolic process;c:Epoxygenase P450 pathway;d:Drug metabolic process;e:Regulation of attachment of spindle microtubules to kinetochore;f:Xenobiotic metabolic process;g:Steroid metabolic process;h:Gluconeogenesis;i:Cellular response to calcium ion;j:Androgen metabolic process.
2.3 Hub基因的分析
构建一个包含112个节点和231连接线的PPI网络(图3A)。然后,使用来自Cytoscape的CytoHubba应用程序识别DEG 的中心基因(图3B)。在这个网络中,得到了10个节点和44连接线;这些节点代表10个基因:细胞周期蛋白依赖性激酶1(CDK1)、细胞周期蛋白B1(CCNBI)、核分裂周期80(NDC80)、拓扑异构酶(DNA)II α 170 000(TOP2A)、上皮细胞转化序列2癌基因(ECT2)、细胞周期相关蛋白激酶2(NEK2)、肌动蛋白结合蛋白(ANLN)、泛素蛋白连接酶同源物(DTL)、核糖核苷酸还原酶M2(RRM2)和透明质酸介导运动因子受体重组蛋白(HMMR)。同时,CDK1、CCNBI和NDC80是基于相互作用得分的前3个优秀基因。
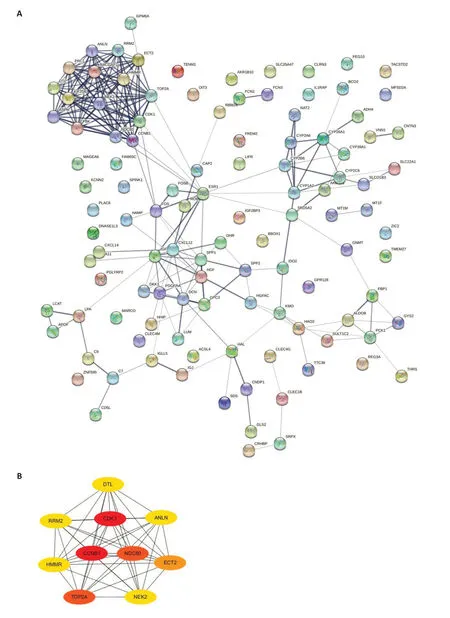
图3 DEGs与前10个关键基因之间的PPI网络构建Fig.3 Protein-protein interaction (PPI) network among the DEGs and the top 10 hub genes.A:PPI network.Circles represent genes,lines represent the interaction of proteins between genes,and the results within the circle represent the structure of proteins.Line colors indicate the interaction between the proteins.B:The most significant module was obtained from the PPI network of DEGs using CytoHubba,including 112 nodes and 231 edges.
2.4 Hub基因在HCC患者中的预后意义
在Wurmbach肝脏数据集中,3个hub基因显著相关(图4A)。根据cBioPortal数据库(图4B~D),3个关键基因的CDK1、CCNB1和NDC80之间存在显著相关性(P<0.05)。Pearson 和spearman 得分均超过0.8。在Wurmbach肝脏数据集中,3个hub基因显著相关(图4A)。根据cBioPortal 数据库(图4B~D),3 个关键基因的CDK1、CCNB1和NDC80之间存在显著相关性(P<0.05)。Pearson和spearman得分均超过0.8。

图4 hub基因与表达的相关性Fig.4 Correlation among the 3 hub genes and their expressions.A-D:Correlations between CDK1,CCNB1 and NDC80 in HCC.
在TCGA数据集中,CDK1、CCNB1和NDC80在肝细胞癌中的mRNA 表达明显高于邻近组织(图5A~C)。此外,Oncomine数据库用于分析正常、肝硬化、肝细胞癌和肝细胞发育不良条件下hub 基因的表达(图5D~F)。正常、肝硬化和肝细胞发育不良之间没有显著差异,但在HCC中表达显著增加。在筛选了3个hub基因后,我们在Kaplan-Meier绘图仪中对目标基因进行了OS分析。结果表明,这些hub基因的突变可能导致HCC 患者的OS较差,表明这些基因具有潜在的诊断价值(图5G~I)。随后,使用Oncomine,在四个不同的数据集中,我们还发现hub基因的表达在肿瘤组织中显著上调(图6A~C)。

图5 hub基因在不同类型肝组织中的表达及CDK1、CCNB1和NDC80的ROC分析Fig.5 Expressions of CDK1,CCNB1 and NDC80 in different types of liver tissues and ROC analysis.A-C:Validation of the expression of the 3 genes at the mRNA and protein levels by the UALCAN database.D-F:Expression of CDK1,CCNB1 and NDC80 in normal,cirrhotic,HCC and hepatic dysplasia tissues in the Wurmbach liver dataset.G-I:Overall survival analyses of the hub genes using Kaplan–Meier Plotter online platform.
2.5 RT-qPCR实验验证
在17例肝癌中,CDK1、CCNB1和NDC80的表达明显高于其配对的邻近组织(P<0.001;图7A~C)。此外,CCNB1与NDC80的表达呈高度正相关(P<0.001),而CDK1与CCNB1的表达与CDK1与NDC80的表达呈中度正相关(P<0.05;图7D~F)。

图7 验证hub基因在临床样本中的表达Fig.7 Verification of the expression of the hub genes in clinical samples of HCC.A:CDK1.B:CCNB1.C:NDC80.***P<0.001 vs adjacent tissues(n=17).D-F:CDK1,CCNB1 and NDC80 expressions levels were highly correlated with one another(n=17).
3 讨论
肝癌的发生是一个复杂的生物学过程[16,17]。近年来,大量生物标志物被用于肝癌的早期诊断,尤其是慢性乙型肝炎病毒(HBV)[18-20],它是HCC的重要病因之一[21,22]。此外,本研究主要针对HBV感染患者,这与以往研究仅针对HCC患者有所不同。尽管一些研究使用了与本研究相似的数据集,但得到了不同的结果。首先,本研究使用“sva”包去除批次效应,减少分析误差,使用不同插件分析hub 基因[23,24],本研究筛选出HBV-通过检查GEO数据库的3个芯片数据集来关联HCC和正常组织。为了避免每个数据集在测量平台和实验室条件下的差异,我们使用“sva”包来消除批间差效应。在371份HBV阳性样本中,与非肿瘤肝组织相比,在HCC组织中共鉴定出121个DEG。KEGG分析表明,DEG富含代谢途径,如视黄醇代谢、咖啡因代谢和药物代谢-细胞色素P450。这些结果表明,DEGs显著影响细胞分裂和代谢途径。氧化还原过程、外源性药物分解代谢过程和纺锤体微管与动力连接的调节是前3个最显著富集的BP。所有这些BP术语在维持生物体的正常生长和代谢方面都发挥着重要作用。
最后,表明10 个基因CDK1、CCNB1、NDC80、TOP2A、NEK2、ECT2、ANLN、DTL、RRM2、HMMR与癌细胞的生长和存活有关。据报道,TOP2A 可以在许多癌症类型中诱导肿瘤的发展和进展。目前大多数研究表明,异常TOP2A表达诱导肿瘤的增殖、转移和化疗耐药等恶性特征主要是通过DNA拓扑状态的调节和复制。此外,TOP2A是一些最广泛使用的用于治疗人类癌症的化疗药物的靶点[25,26]。NEK2通过其底物C-NAP1的可逆磷酸化调节有丝分裂中心体分离,在维持中心体完整性方面发挥着重要作用。癌细胞中过度暴露NEK2导致CIN、细胞增殖和耐药性增强[27,28]。最近的一项研究还表明ECT2与早期复发性HCC疾病和较差的生存率显著相关。敲除ECT2可显著抑制Rho家族的小分子鸟苷酸三磷酸酶(Rho GTPases)的活性,促进细胞凋亡,减弱致癌性并降低HCC细胞的转移能力[29,30]。ANLN缺乏诱导多核细胞数量增加,同时激活细胞凋亡信号和DNA损伤检查点。此外,HBV感染通过抑制microRNA(miR)15a和miR 16 1的表达增加了ANLN的表达,这两个都通过靶向其3个非翻译区而被鉴定为ANLN 上游阻遏物。ANLN 通过减少细胞凋亡和DNA损伤的方式促进肿瘤生长。ANLN的表达水平显著影响HCC患者的生存概率,可能代表一个有前景的预后生物标志物[31]。据报道,靶向DTL减少了细胞周期调节因子和染色体分离基因,导致细胞微核化增加。DTL耗竭抑制肝癌细胞生长、增加衰老和减少肿瘤发生[32,33]。据报道,RRM2是索拉非尼的新靶点,部分有助于其在HCC细胞中的抗癌活性[34]。
KEGG分析显示,“视黄醇代谢”、“咖啡因代谢”和“p53信号通路”的富集程度最高,表明DEGs显著影响细胞分裂和代谢通路,促进晚期复合物依赖性分解代谢过程、氧化还原过程和细胞是前3个具有最显著富集的BP。肿瘤发生的主要原因可能是细胞周期失衡,导致细胞过度增殖。以前的研究报道,HBV感染可引起代谢信号通路的变化。结果可能会改变正常的肝细胞代谢并促进HBV相关HCC的进展。
除上述关键基因外,本研究重点关注CDK1、CCNB1和NDC80基因。CDK1与较差的HCC 总生存率相关(P=0.008)。CDK1高表达是1年和5年肿瘤复发的危险因素(P=0.013和P=0.017),表明CDK1可能在HCC进展中发挥重要的致癌作用[35]。CDK1是Serd/Thr 蛋白激酶的成员,对细胞G1/S和G2/M相变至关重要[36-38]。机制研究表明FOXM1直接结合CCNB1的启动子区域并调节表达水平CCNB1 基因的转录水平。CCNB1的高表达与HCC患者的不良预后密切相关[39-41]。在既往研究中,HCC组织中NDC80的表达水平显著高于邻近组织。NDC80的组合式导致细胞凋亡增加和S期细胞周期停滞。NDC80通过减少细胞凋亡和克服细胞周期停滞来促进HCC 进展[42]。在本研究中,使用TCGA数据的进一步分析表明CDK1、CCNB1和NDC80之间存在密切相关性。HBV相关肝癌中3个hub基因的表达明显高于邻近组织。肝癌患者的生存率显著相关。CDK1、CCNB1和NDC80在HCC组织中的高表达与预后不良和高复发风险有关。然而,这3个基因在肝癌和其他癌症类型中的生物学功能,包括代谢和细胞周期,需要进一步研究。这可能表明它们作为治疗肝癌或其他癌症类型的靶标的效用,并提供有关它们相互作用的详细信息。
总之,通过基因组水平和生物信息学分析,本研究确定了与肝癌发生有关的DEGs,同时,PPI网络也揭示了DEGs在这些通路中的相互作用,并最终确定了3个基因标志物。只有少数研究集中在3个基因之间的联系,本研究试图做到这一点,一些结果与先前的研究一致,但NDC80与肝癌的关系鲜少被报道,以后的研究应该重点关注。虽然这些数据集在之前的研究中已经部分使用,但本研究不同于之前的研究[10,23,43,44],本文综合运用了3个数据集,而不是直接线上分析,相比之前的研究,增加了R包去除批间差效应,大大降低了外界因素导致的分析误差,而且,此研究不仅仅停留于分析层面,并收集临床样本进行验证,对采集的17对临床样本进行了RT-qPCR实验验证,结果表明与分析一致。本研究为有HBV感染史的患者提供了HCC的新病因和非肿瘤肝组织转化为HCC组织的分子机制。重要的是,这些结果可能为这些患者的靶向治疗提供一些潜在的治疗靶点,有助于肝癌的早期诊断和治疗。
猜你喜欢
皮肤性病诊疗学杂志(2021年5期)2021-11-27
现代临床医学(2021年4期)2021-07-31
昆明医科大学学报(2021年4期)2021-07-23
云南医药(2021年3期)2021-07-21
三农资讯半月报(2021年1期)2021-01-27
天津医科大学学报(2021年1期)2021-01-26
中西医结合肝病杂志(2020年2期)2020-10-27
中国医药导报(2016年33期)2017-03-06
医学研究杂志(2015年12期)2015-06-10
癌变·畸变·突变(2015年4期)2015-02-27
