
碳包覆合金/过氧化氢非均相Fenton体系高效矿化全氟辛酸*
2021-11-09 11:53资凯禄董方雅田凯勋
湘潭大学自然科学学报 2021年4期
资凯禄, 董方雅, 田凯勋, 沈 健*, 吴 达
(1. 湘潭大学 环境与资源学院,湖南 湘潭 411105;2.华北制药华胜有限公司,河北 石家庄 052160)
0 引言
全氟羧酸类化合物(Perfluorinated carboxylic acids,PFCAs)因其具有优良的热稳定性、化学稳定性和疏水疏油特性,被广泛应用于日常用品和化学生产[1-3].然而,作为一类人工合成的新型持久性有机污染物,PFCAs能够引起水体污染并对生物体具有“三致”效应,其中,全氟辛酸(Perfluoro octanoic acids, PFOAs)是最为常见、最典型且商业用途最多的一种全氟羧酸类化合物[4-9].
基于PFOAs环境生态不兼容性,现有水处理技术目的是将其完全从环境中去除.目前,去除PFOAs的方法主要有吸附、生物降解、高级氧化法等[10-15].虽然每种方法能够一定程度地去除PFOAs,但是也存在各自的缺陷.例如,吸附法脱除PFOAs主要通过污染物转移,未将其彻底去除,仍需要后续深度处理;生物降解周期过长、PFOAs去除率不高,并且对微生物有一定的毒害作用;高级氧化包括光催化、电催化、热处理、Fenton氧化,其中,光、电和热催化降解可实现PFOAs矿化,但矿化效率和能量有效利用率低.相对于光、电和热催化技术,Fenton氧化法具有操作简单、反应条件温和等优点,并且已广泛应用于难生化、高毒性的有机废水深度处理.众所周知,Fenton氧化反应主要依靠过氧化氢和富电子(如Fe2+、Fe0、碳材料等)催化剂,产生具有强氧化性的羟基自由基(·OH),使废水中的有机物裂解矿化,最终生成二氧化碳和水.对于PFOAs降解体系,·OH难以打破C-F键,而超氧自由基和过氧化氢自由基能够实现C-F断裂,并矿化PFOAs,因此,导致传统Fenton降解PFOAs效率极低.此外,Fenton反应需要在强酸环境下发生,使得催化剂易于溶出,催化剂的活性和稳定性显著降低.
针对上述问题,本文拟通过制备碳包覆金属复合催化剂,缓解金属浸出,保持催化剂活性和稳定性.同时,利用碳材料与金属之间界面应变,促进电子迁移,调控活性自由基产生路径,提高PFOAs降解效率.因此,本文采用类金属有机框架材料为前驱体,在高温条件下,利用有机配体碳化与金属还原的速率差异性,获得碳包覆金属催化剂.采用透射电子显微镜(Transmission electron microscope, TEM)、X射线衍射仪(X-ray diffraction,XRD)、N2吸脱附等温仪、振动样品磁强计(Vibrating sample magnetometer,VSM)表征催化剂形貌和结构,采用X射线光电子能谱(X-ray photoelectron spectroscopy,XPS)探究催化剂微观电子结构和潜在电子转移途径,并结合总有机碳分析仪(Total organic carbons,TOC)和电子顺磁共振分析仪(Electron paramagnetic resonance,EPR)阐述潜在的PFOAs降解机制.
1 实验
1.1 材料与试剂
六水合硝酸镍(分析纯,≥98.0%,广东光华科技股份有限公司)、氢氧化钠(分析纯,96.0%,天津市大茂化学试剂厂)、盐酸(分析纯,36.0%~38.0%,天津市大茂化学试剂厂)、硝酸钴(分析纯,99%,Aladdin)、铁基金属有机框架材料R-Fe(自制,美国西北太平洋国家实验室)、全氟辛酸(分析纯,90%,Aladdin)、无水乙醇(分析纯,≥99.7%,湖南汇虹试剂有限公司)、过氧化氢(≥30.0%,西陇科学股份有限公司)、高纯氩气(≥99.999%,长沙日臻气体有限公司).实验用水均为超纯水.
1.2 实验设备与分析仪器
TEM(FEI TECNAI G20,Field Electron and Ion Company,美国)、XRD(*/D/MAX-2500/PC,日本理学)、N2吸脱附等温仪(*/ASAP2020PLUS HD88,美国麦克仪器公司)、VSM(PPMS-9,Quantum Design)、XPS(ESCALAB 250 XI,赛默飞)、TOC(TOC-LCPH,岛津(香港)有限公司)和EPR(布鲁克A300).
1.3 催化剂合成方法
采用共沉淀法合成.分别在100 mL去离子水中加入0.010 mol R-Fe和0.018 mol Co(NO3)2·6H2O溶液.在搅拌速度为300 r/min下,向硝酸钴溶液中匀速逐滴加入R-Fe溶液并混合,陈化24 h.用去离子水洗涤固体沉淀物,直到溶液pH值为中性,再用乙醇洗涤3次.最后,所得产品在常压和50 ℃条件下干燥12 h[16].将干燥后材料在高纯氩气氛围下,以5 ℃/min升温至600 ℃,保持2 h,自然冷却至室温,收集至试剂瓶中密封保存,分别记为C@FeCo.
1.4 降解实验过程
本论文主要通过考察H2O2用量、pH、催化剂用量优化PFOAs降解性能.具体实验过程如下(以调控H2O2用量为例):首先,用去离子水配置30 mg/L的PFOAs溶液,用去离子水配置1 mol/L的HCl溶液;其次,用移液管量取30 mL PFOAs溶液至样品瓶中(共5组),用HCl溶液调节pH=2,分别称取30 mg C@FeCo催化剂至溶液中,在五个样品瓶中分别加入5、10、20、25、30 mmol/L的H2O2溶液,在25 ℃下匀速搅拌(200 r/min)反应2 h;待反应结束后,水样用0.45 μm滤头过滤,倒入离心管中,置于 4 ℃冰箱中保存;对样品进行TOC测试并计算PFOAs矿化率(以TOC去除率为计).考察pH和催化剂用量需在上述实验过程中相应部分做修改.
2 结果与讨论
2.1 形貌与结构表征
TEM图显示出C@FeCo催化剂形貌、元素分布和晶格间距等特性与参数,如图1所示.由图1(a)可见,所制备C@FeCo分散较均匀,并且碳膜均匀地包裹在FeCo表面,可认为所获得C@FeCo催化剂为碳包覆结构.根据XRD谱图(图1(b)),C@FeCo衍射角(2θ)在44.919°、65.360°和82.840°处分别代表FeCo(110)、(200)和(211)晶面特征峰,且在FeCo(110)晶面处的衍射峰强度最高.通过计算,所得各特征峰的晶面间距分别为0.201 8 nm、0.142 8 nm和0.116 53 nm.结合图1(a),FeCo晶面间距为0.20 nm,与XRD谱图中FeCo(110)吻合.因此,利用有机配体碳化和金属还原速度的差异性,可以获得具有碳包覆金属核壳结构的催化剂.氮气等温吸脱附曲线和孔径分布曲线能够清晰地反映材料的孔径信息和尺寸.由图1(c)和1(d)可知,孔径分布曲线显示C@FeCo材料的孔隙绝大多数都集中在50 nm以下,且氮气吸脱附曲线呈H3型,存在清晰的介孔回滞环,因此,C@FeCo中碳膜为介孔材料.同时,C@FeCo比表面积为74.357 m2/g,有助于污染吸附和过氧化氢活化.对于FeCo相, FeCo通常呈现出典型的软磁性材料特性,即高饱和磁感应强度(160~220 emu/g)和较低的矫顽力(小于100 Oe).然而,根据磁滞回滞曲线(图1(e)),C@FeCo饱和磁感应强度接近于150 emu/g.考虑到C@FeCo中非磁性碳质量百分比约为9%,因此,FeCo饱和磁感应强度真实值约为164 emu/g,满足软磁材料特性.而矫顽力受控于磁晶各向异性能,传统软磁材料晶格结构为各向同性,因此矫顽力较低.对于C@FeCo,其矫顽力为416.66 Oe,意味着FeCo晶格结构由各向同性朝着各向异性转变,增加其磁晶各向异性能.因此,可推测FeCo晶格畸变是FeCo与碳膜的晶格不匹配导致的.
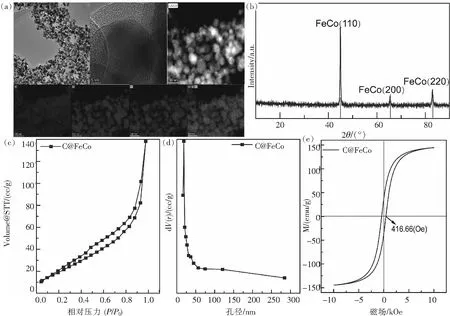
图1 (a) C@FeCo透射电子显微镜图(低倍电镜、高倍电镜、EDS-HAADF和元素分布图),(b)X射线衍射图, (c)氮气吸脱附曲线,(d)孔径分布图和(e)磁滞回滞曲线Fig.1 (a) TEM images (Low-resolution TEM, High-resolution TEM, EDS-HAADF image, EDS mappings), (b) XRD pattern,(c) Nitrogen adsorption-desorption profiles, (d) Pore size distributions, and (e) M-H loop of C@FeCo
2.2 电子结构
XPS可以反映材料的元素组成和价态.如图2(a)所示,C@FeCo C1s结合能在284.8 eV、286.4 eV、289 eV分别代表C-C键、C-O键和C=O键,C-C键、C-O键、C=O键分别占比80.52%、13.82%、5.66%.与标准C1s对比,C@FeCo的C-C键结合能向低处偏移,意味着其电子密度增大.图2(b)表明,Co 2p谱图中结合能为779.7 eV、780.5 eV、794.9 eV和796 eV,分别对应Co02p3/2、Co2+/Co3+2p3/2、Co02p1/2、Co2+/Co3+2p1/2.同样对于Fe2p谱图,结合能为708.2 eV、711.2 eV、713.6 eV、721 eV、724.1 eV和726.3 eV,分别对应Fe02p3/2、Fe2+2p3/2、Fe3+2p3/2、Fe02p1/2、Fe2+2p1/2、Fe3+2p1/2.与标准Co2p和Fe2p对比,零价Co元素附近电子密度减少,零价、二价和三价Fe元素的结合能均向高处偏移,电子密度减小.结合图1(d)分析、推测催化剂FeCo相可能在高温热处理过程中发生晶格扭曲,使得FeCo与碳膜界面处不匹配程度增加,导致Fe元素和零价钴附近的电子密度降低,C-C键和零价钴附近的电子密度增加.
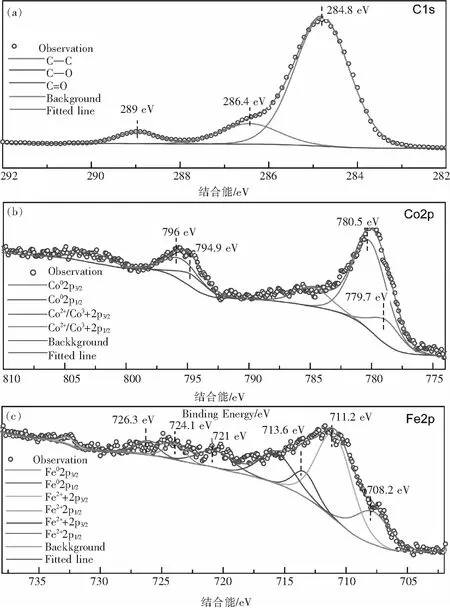
图2 C@FeCo X射线光电子能谱图(a)C1s,(b)Co2p和(c)Fe2pFig.2 XPS spectrums of C@FeCo (a) C1s, (b) Co2p, and (c) Fe2p
2.3 降解性能
为研究H2O2浓度对PFOAs降解性能的影响,在30 mL 30 mg/L的PFOAs溶液、pH=2、催化剂用量1 g/L、反应温度25 ℃的条件下,H2O2的浓度分别为5、10、20、25、30 mmol/L,反应2 h,观察两种催化剂在不同H2O2浓度条件下TOC去除率的变化,如图3(a)所示.由图3(a)可知,C@FeCo催化剂,随着H2O2浓度的升高,PFOAs的TOC去除率呈现先降低再升高的趋势,即在H2O2浓度为30 mmol/L时,PFOAs TOC去除效率最高,为79.72%,优于现有Fenton工艺效果.在H2O2的最佳浓度条件下, pH值和催化剂投加量对PFOAs降解性能也有直接影响.通过改变不同的pH和催化剂投加量,发现C@FeCo催化剂在pH为2且投加量为1 g/L的条件下,TOC去除率达到最佳(图3(b)和3(c)).因此,C@FeCo催化剂降解PFOAs的最佳条件是pH为2,催化剂用量为1 g/L,H2O2投加量为30 mmol/L.

图3 H2O2投加量(a)、pH(b)和催化剂投加量(c)对PFOAs矿化率影响Fig.3 Effects of H2O2 dosage (a), pH (b), and catalysts dosage (c) on mineralization efficiency of PFOAs
2.4 潜在构效关系
如前所述,C@FeCo具有较好的PFOAs催化降解性能.TEM和VSM分析结果表明,C@FeCo催化剂在热退火过程(碳层包覆过程)中发生了较大程度的晶格扭曲,导致晶格界面的不匹配,进而导致合金相与碳层间产生界面应变.同时,XPS进一步阐述了电子在界面处迁移.为研究界面应变效应诱导电子迁移过程对过氧化氢活化和活性自由基产生路径的影响,采用EPR进一步分析活性自由基,如图4所示.根据图4峰强度可看出,HO·峰强度最弱,其次为超氧阴离子(O2-·),过氧化氢阴离子(HO2-)强度最强.先前研究表明:HO2-降解PFOAs效率最高,最高可达80%,其次为O2-·可去除~60% PFOAs,而HO·对PFOAs去除几乎无效.因此,C@FeCo催化剂高效降解PFOAs很大程度归因于O2-·和HO2-强氧化作用.而不同于传统Fenton体系H2O2活化路径,基于界面应变的作用更有利于形成富电子碳膜层,该碳膜有助于活性氧化物种(O2-·和HO2-)形成.
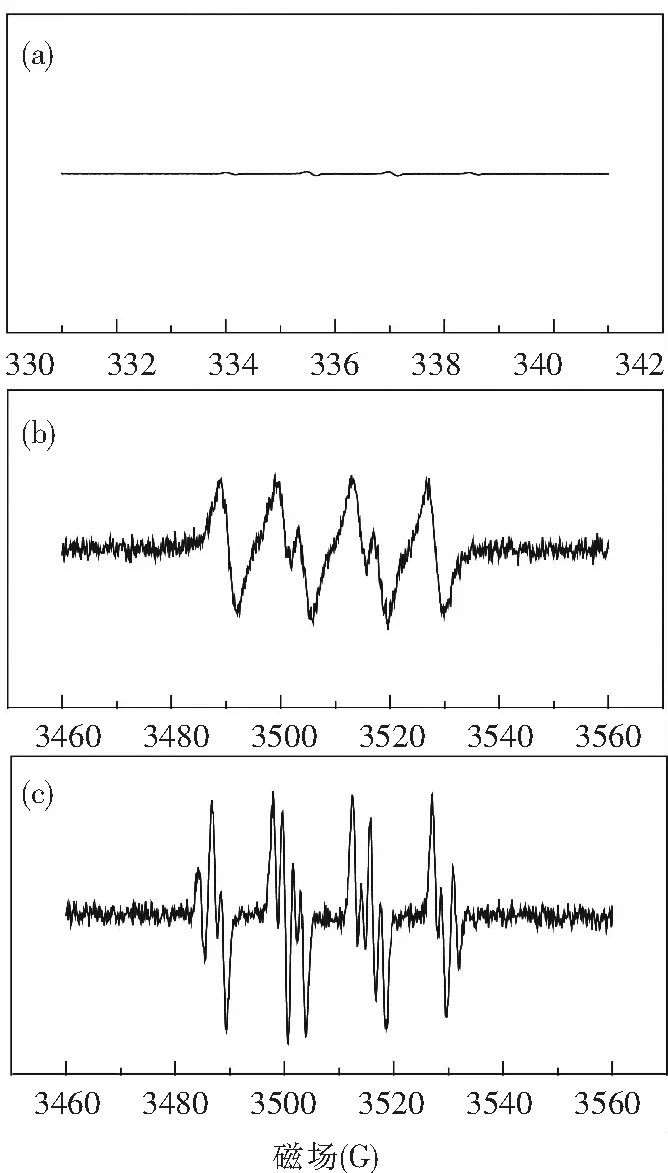
图4 EPR谱图(a)HO·,(b)O2-·和(c)HO2-Fig.4 EPR spectrums (a) HO·, (b) O2-·, and (c) HO2-
3 结 论
(1) C@FeCo催化剂在pH=2,反应温度25 ℃,催化剂用量1 g/L,H2O2用量30 mmol/L的条件下,PFOAs矿化率(以TOC去除率计)最高,为79.72%.
(2) PFOAs高矿化率是以下原因导致:利用金属相与碳层之间晶格不匹配导致的晶格扭曲,促进电子向碳膜迁移,进而改变过氧化氢活化路径,产生超氧自由基和过氧化氢自由基,从而实现高效降解PFOAs.
猜你喜欢
工业安全与环保(2022年10期)2022-10-28
山东冶金(2022年3期)2022-07-19
浙江大学学报(理学版)(2020年1期)2020-03-12
数学物理学报(2019年5期)2019-11-29
数学物理学报(2017年5期)2017-11-23
武大国际法评论(2016年2期)2016-06-01
潍坊学院学报(2016年6期)2016-04-18
天津城建大学学报(2015年5期)2015-12-09
安徽农学通报(2015年2期)2015-02-12
长江大学学报(自科版)(2014年1期)2014-03-20
