
通过微量细胞鉴定快速生产基因编辑猪*
2021-11-05 03:00王娇祥范柠粼施德佳陈姝含王璐璐李莲军李鸿辉魏红江
云南农业大学学报(自然科学) 2021年5期
王娇祥,范柠粼,施德佳,陈姝含,王璐璐,李莲军,李鸿辉,魏红江
(1.云南省动物基因编辑与体细胞克隆技术重点实验室,云南 昆明 650201;2.云南省异种器官移植工程研究中心,云南 昆明 650201;3.云南农业大学 动物医学院,云南 昆明 650201;4.云南农业大学 动物科学技术学院,云南 昆明 650201)
体细胞克隆羊“Dolly”的诞生首次证明了高度分化的成体细胞可通过卵母细胞质重编程后获得胚胎发育的全能性及成活的个体[1],2000 年体细胞克隆技术在猪上取得了重要突破[2],随后基于同源重组技术首次实现猪成纤维细胞基因编辑,并结合体细胞克隆技术获得了α-1,3-半乳糖基转移酶(alpha-1,3-galactosyltransferase,GGTA1)敲除克隆猪[3]。近年来CRISPR/Cas9 基因编辑技术在哺乳动物基因组编辑中得到了广泛应用,结合体细胞克隆技术已先后获得多种不同基因修饰的克隆猪,实现了猪新品种培育[4]、人类疾病模型构建和发病机制研究[5]。因此,猪的体细胞核移植(somatic cell nuclear transfer,SCNT)在生物医学和比较医学的发展中具有巨大的潜力。
尽管原核显微注射[6]、慢病毒[7]和精子介导的基因传递以及胞浆内精子注射[8]等技术已在动物基因编辑中取得进展,但是这些技术产生的基因编辑动物通常效率低下且为嵌合体。体细胞基因编辑结合SCNT 产生的基因编辑猪基因型明确且可遗传给后代,是生产基因编辑猪的首选方法。但是,猪成纤维细胞的增殖能力有限,长时间培养后更容易老化[9]。同时,多基因编辑和长期细胞筛选过程造成猪基因编辑阳性细胞DNA 损伤或突变,影响基因编辑克隆猪的生产效率。因此,快速有效地筛选基因编辑阳性供体细胞系是获得基因编辑克隆猪的重要前提。
在筛选基因编辑阳性细胞的过程中,首先利用构建成功的目的基因打靶载体转染成纤维细胞,然后通过极度稀释培养出单细胞克隆或通过流式细胞术分选后进行单细胞培养,这通常需要添加相应的抗性药物进行长期培养和筛选,直至单细胞克隆形成。该筛选过程可能对细胞造成物理损伤或化学毒性,减慢细胞的生长速度,甚至引起DNA 损伤或突变[10]。有研究报道一些单细胞克隆的形成长达22 d[11-12],鉴定阳性基因编辑细胞系所需的细胞数量高达103个[11,13],导致大量细胞衰老,可供传代的细胞数量减少,甚至筛选出的阳性细胞无法继续传代而死亡,严重影响基因编辑克隆猪产生的效率。考虑到猪基因编辑阳性细胞的筛选周期长、筛选效率低和成功率低的问题,本研究利用已知基因编辑阳性细胞系建立微量细胞鉴定阳性细胞克隆的方法,然后通过鉴定未知的基因编辑阳性细胞系证明该方法的可行性,缩短细胞筛选周期,提高猪基因编辑阳性细胞系的筛选效率,对快速生产基因编辑猪具有重要的参考价值。
1 材料与方法
1.1 试验材料
本研究所用的野生型滇南小耳猪胎儿成纤维细胞来自云南农业大学实验动物中心,卵巢采自云南省昆明市呈贡区鸿腾屠宰场。所有试剂耗材除特别说明外均购自Gbico、Corning、Sigma-Aldrich、BI、Solarbio、Ausbian 和Lonza 公司。所有的试验均在云南农业大学云南省动物基因编辑与体细胞克隆技术重点实验室完成。
1.2 基因编辑猪成纤维细胞系的梯度计数
将基因编辑的滇南小耳猪成纤维细胞系进行传代培养,待细胞生长至40%~60%的汇合度时,用0.25%的胰蛋白酶消化后置于显微操作系统,按20、50、100、150 和200 个细胞计数后分别转移至含2 μL 细胞裂解液的PCR 管中用于基因型鉴定,每个梯度3 个重复。
1.3 细胞基因型鉴定并确定鉴定所需最低细胞量
将计数的20、50、100、150 和200 个细胞用细胞裂解液分别裂解后提取细胞基因组DNA,进行目的基因片段的PCR 扩增、T7EN1酶切和Sanger 测序,然后进行琼脂糖凝胶电泳。利用Image Lab 软件(Bio-Rad v6.0)进行电泳条带的灰度值分析,测序结果利用SnapGene 软件(GSL Biotech v3.2.1.0)进行序列比对,确定细胞基因型鉴定所需的最低细胞量。
1.4 基因靶向载体的转染和单细胞克隆培养
为验证微量细胞鉴定方法的可行性,我们设计了基于CRISPR/Cas9的基因编辑载体来靶向猪IPO13基因(sgRNA1:ACATCAAGATCTCTCGCTAT;sgRNA2:GCAGCTACTGCAGCCCGACA)。转染前2 d,将野生型滇南小耳猪胎儿成纤维细胞解冻并传代。当细胞达到40%~60%汇合度时,用0.25%胰蛋白酶消化。然后加入2%FBS 终止消化,并将细胞在室温下以1 200 r/min离心3 min,收集细胞沉淀并准确计数。取大约3×105PFF,再次离心,收集沉淀。将载体(mCas9:msgRNA=2∶1)与电穿孔缓冲液充分混合。将细胞温育5 min,充分混合并转移至电击杯中,在设定的Lonza-4D 核转染仪程序下进行转染。电击完成后,加入约80 μL 完全细胞培养液,将电击杯置于5% CO2、38 ℃培养箱中静置15 min,轻轻将细胞转移至加有完全培养液的6 cm 培养皿中继续培养。48 h 后,加入2.0 μg/mL的 Puromycin 维持筛选2 d 后,极度稀释至10 cm 培养皿进行单克隆培养,每3 d 换液1 次,约9 d 时单细胞克隆形成,在显微镜下用记号笔圈出单克隆,弃除上清液,PBS 清洗2 遍,将单细胞克隆用0.25%胰蛋白酶消化后,挑取单细胞克隆于96 孔板继续扩大培养,选取细胞生长汇合度为70%~80%的孔消化,取20 μL 细胞悬液准确挑取既定数量的细胞至PCR 管中,进行后续的细胞基因型鉴定。
1.5 基因编辑单细胞克隆的基因型鉴定
将既定数量的细胞裂解后提取基因组DNA作为模板,利用特异性引物(IPO13-F:5′-AAGCACTTGAGCACCTTCTGAC-3′;IPO13-R:5′-AAGTTCAGGCTCTCAAGACATC-3′)进行PCR。使用PCR 纯化试剂盒(AP-PCR-50,Axygen,New York,USA)纯化PCR 产物,进行T7EN1酶切和Sanger 测序。将阳性细胞克隆所对应的PCR 产物通过TA 克隆到pMD19 T 载体上,挑取8 个单克隆细菌菌落进行Sanger 测序。双等位基因缺失所对应的细胞克隆选为供体细胞进行扩大培养,用作后续的SCNT。
1.6 胎儿成纤维细胞的分离与提取
手术获得35 d的基因编辑胎儿,立即将其浸入含5%青霉素—链霉素溶液(PS)和10% FBS的DMEM 溶液中,并运送到实验室。在层流罩下去除胎儿的头、尾、四肢和内脏,用无菌PBS(含5% PS)将剩余的胎儿组织清洗3 次,并用不含PS的PBS 清洗5 次。用眼科剪刀将组织切成碎片,转移至T25 烧瓶中,加入3 mL 胶原酶IV,并将组织在37 ℃恒温箱的水平摇床上消化1 h。通过离心除去胶原酶后,将收集的细胞在37 ℃、5% CO2的培养箱中用含有2% FBS 和1%PS的DMEM 培养基培养。每3 d 更换1 次培养基,传代5×104细胞/mL的密度并冷冻保存备用。
1.7 体细胞核移植和胚胎移植
体细胞核移植按已有报道的方法[14]进行,即将体外成熟培养的卵母细胞利用显微操作系统去核后,再将基因编辑阳性细胞注射到去核的卵母细胞中,经过电融合和电激活后,移入PZM-3 培养液中进行体外培养获得重构胚胎。将重构胚移入至发情代孕母猪的输卵管内,移植后29~33 d时通过B 超(HS-101V,Japan)检测代孕母猪的妊娠情况,确定妊娠后35 d 通过外科手术获取IPO13KO胎儿。参照1.5 节的方法鉴定胎儿的基因型。以确定为IPO13基因编辑阳性的胎儿成纤维细胞作为供体细胞,进行第2 轮体细胞核移植和胚胎移植,通过B 超检测代孕母猪妊娠情况,妊娠约114 d 时代孕母猪分娩获得IPO13克隆仔猪。用同样的方法鉴定IPO13克隆仔猪的基因型。
1.8 统计分析
PCR 和T7EN1的定量数据以“平均值±标准差(mean±SD)”表示;使用SPSS 22.0的student-t检验进行统计分析,包括PCR 结果、T7EN1 以及50CIM (50 cells identified method)和GM (general method)筛选时间。使用校正后的四格表卡方检验比较50CIM 和GM 筛选的成功率。P<0.05表示差异显著,P<0.01 和P<0.001 表示差异极显著,并使用GraphPad Prism 7 软件制图。
2 结果与分析
2.1 最小数量基因编辑阳性细胞的确定
为确定用于基因型鉴定所需的最小细胞量,利用发表的已知基因型的基因编辑阳性细胞系(GHRKO)[15]来建立方法。首先,计数不同细胞数(图1a),然后进行PCR 扩增,结果显示:各细胞组扩增出的PCR产物都清晰可见(图1b、d),但T7EN1 消化后扩增出的PCR 产物在20 个细胞组难以辨清,在其他组中均显示出2 条清晰的切割条带(175 和555 bp)(图1c),灰度值分析显示:20 个细胞组与其他组间均存在显著差异(图1e)。由此初步确定50 个细胞为可用于基因型鉴定的细胞量。另外,将最小细胞数(50 个细胞)的PCR 产物进行TA 克隆和测序,GHR基因带有47 和46 bp的缺失(图1f),因此认为50 个细胞可用于基因型鉴定。
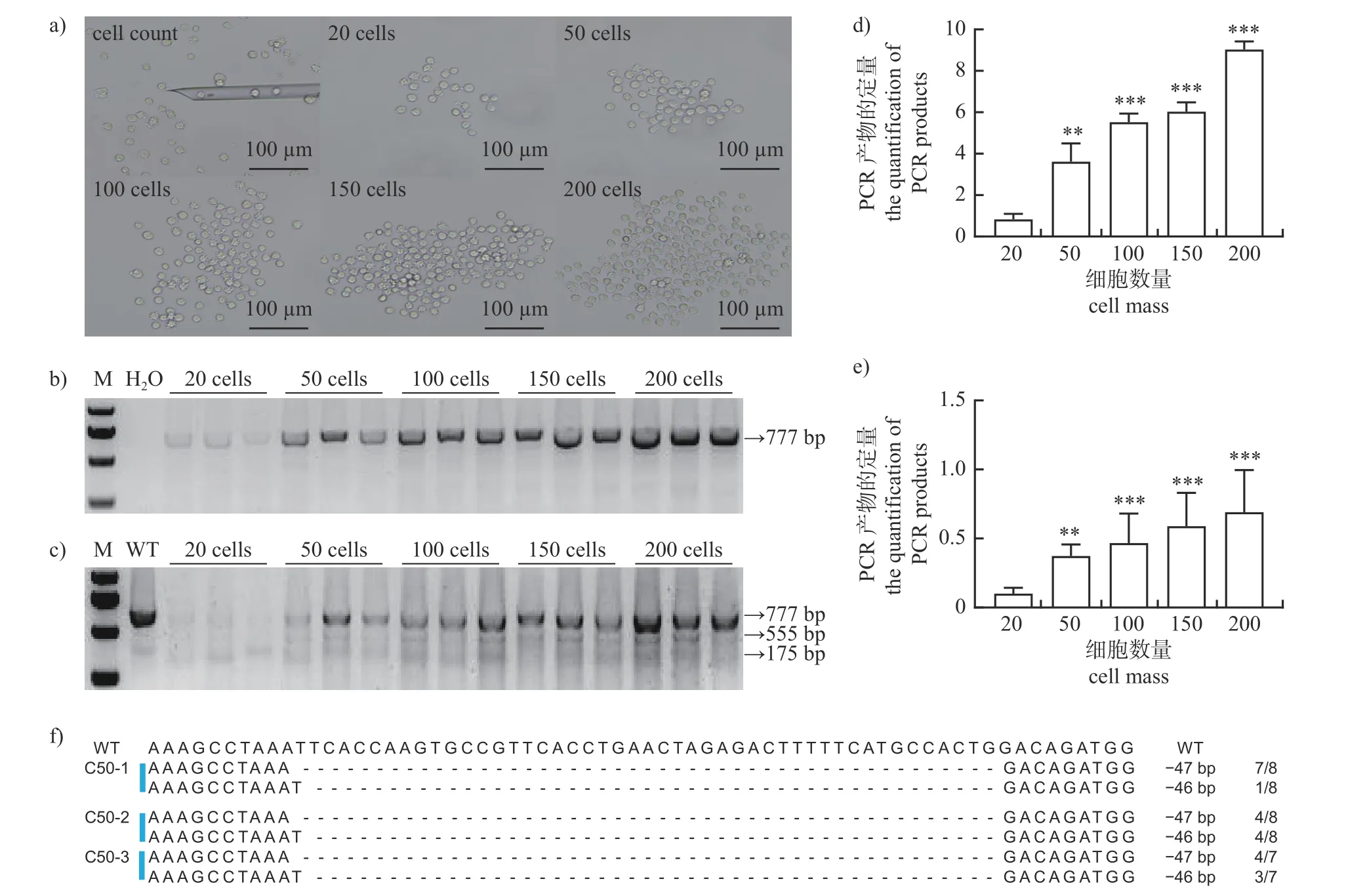
图1 已知不同细胞量的基因编辑阳性细胞基因型鉴定结果Fig.1 Genotype identification results of gene editing positive cells with different cell mass
2.2 最小细胞数用于基因型鉴定的验证
为验证最小细胞数用于基因型鉴定的可行性,我们设计了2 条靶向IPO13KO基因2 号外显子的sgRNA,2 个靶点的距离约为60 bp (图2a)。基因靶向载体转染和单细胞克隆结果表明:4 号和17 号单细胞克隆为基因编辑阳性细胞系(图2b~c);4 号单细胞克隆为IPO13单等位基因缺失细胞系,其中一条链缺失56 bp,另一条链为WT;17 号单细胞克隆为IPO13双等位基因缺失细胞系,其中2 条单链分别发生6 和8 bp的碱基缺失(图2d~e)。该结果也提示在4 号单细胞克隆中,2 条sgRNA 发挥作用但只产生单等位基因缺失;在17 号单细胞克隆中,仅1 条sgRNA(IPO13-sgRNA2)发挥作用但产生了双等位基因缺失。因此,选17 号单细胞克隆为供体细胞,进行后续的SCNT。
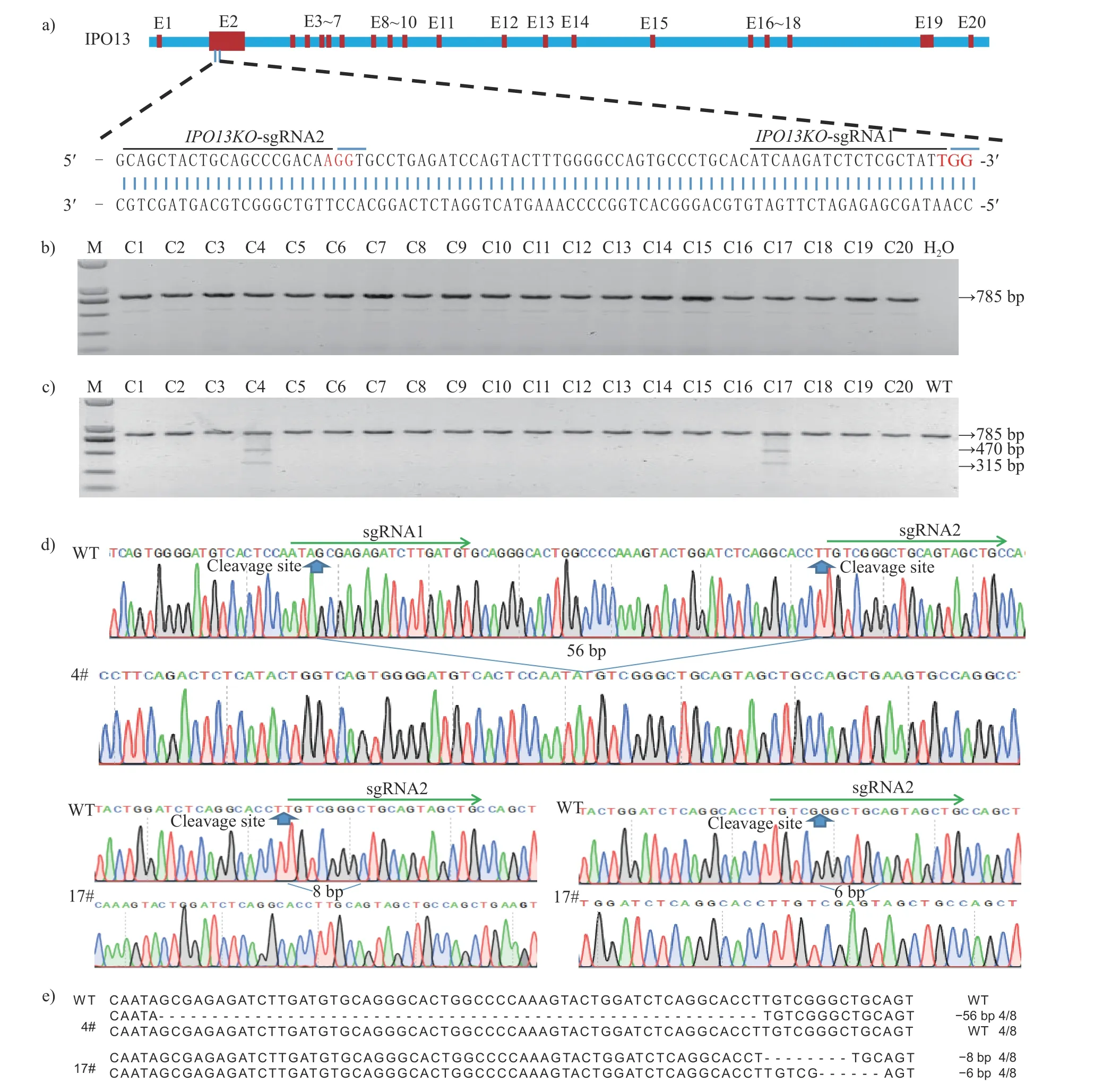
图2 猪IPO13KO 基因编辑阳性单细胞克隆的筛选Fig.2 Selection of porcine IPO13KO positive single-cell colonies
2.3 IPO13KO 胎儿和仔猪的产生
以17 号细胞系作为供体细胞进行SCNT,在第1 轮SCNT 中共构建了1 753 枚克隆胚,并将其移植至5 头代孕母猪中,B 超检测发现有2 头代孕母猪妊娠,妊娠率为40%,其中1 头代孕母猪在妊娠35 d 时通过外科手术获得8 个成活的胎儿。建立胎儿成纤维细胞系,以1 号胎儿成纤维细胞为供体细胞进行第2 轮SCNT,共构建了3 260 枚克隆胚,将其移植至11 头代孕母猪中,B 超检测发现有6 头代孕母猪妊娠,妊娠率为54.5%,4 头分娩,共获得9 头克隆仔猪,6 头成活(表1)。
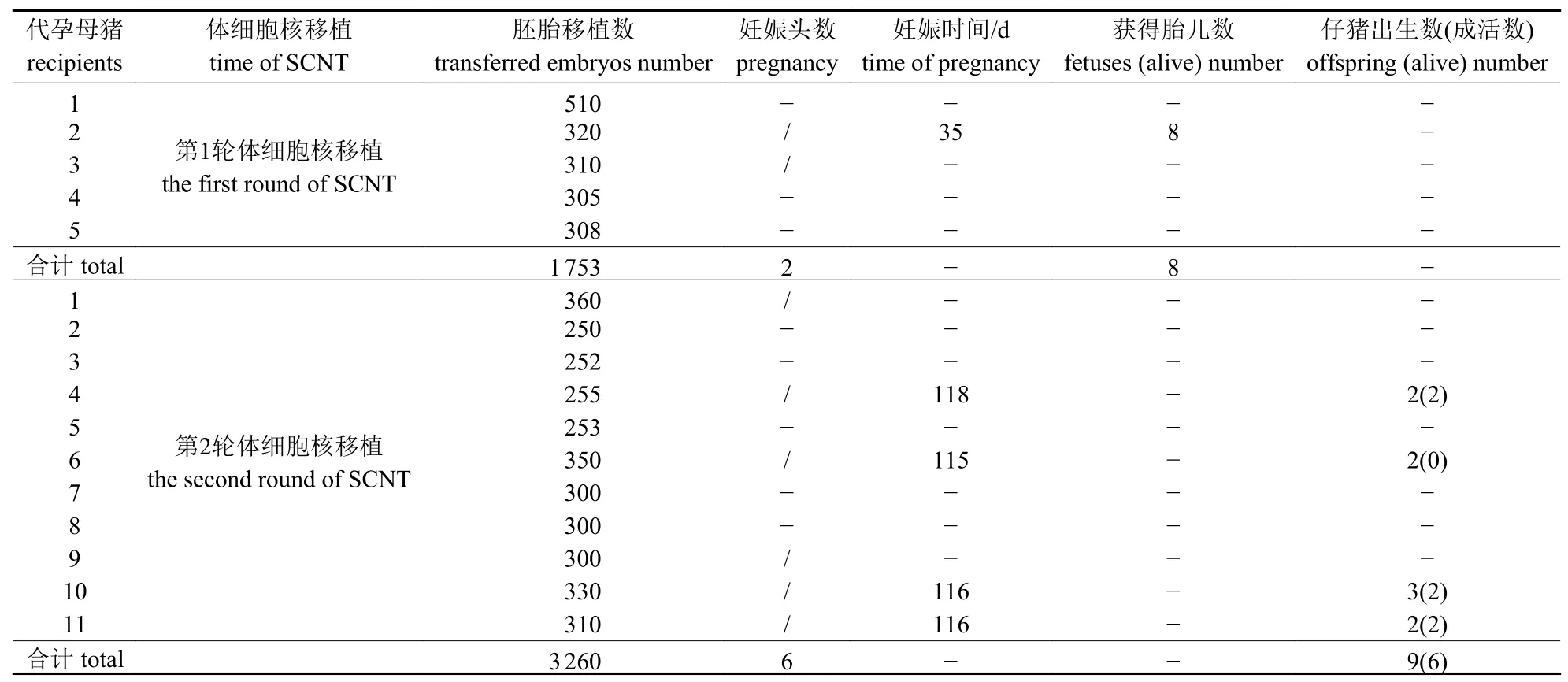
表1 猪IPO13KO 克隆胚胎移植至代孕母猪中的发育情况Tab.1 Development of reconstructed IPO13KO cloned embryos after transfer to recipients
2.4 IPO13KO 胎儿及仔猪基因型鉴定
结果(图3a~c)显示:8 个胎儿的IPO13KO基因片段PCR 产物均为785 bp,进一步通过T7EN1 酶切后分别可见470 和315 bp的分裂条带。Sanger 测序结果显示:在IPO13KO基因靶点的位置分别可见8 和6 bp的碱基缺失,证明所获得的胎儿基因型与17 号供体细胞的基因型完全一致(图3e)。同样,9 头仔猪PCR 产物也为785 bp,T7EN1 酶切后分裂条带为470 和315 bp,Sanger 测序结果为8 和6 bp的碱基缺失,证明所获得的克隆仔猪基因型与17 号供体细胞及克隆胎儿成纤维细胞的基因型完全一致(图3d~e)。
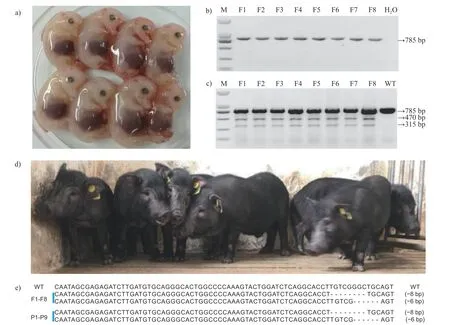
图3 克隆胎儿及仔猪IPO13KO 基因型鉴定Fig.3 Detection of IPO13KO genotypes in fetuses and piglets
2.5 猪基因编辑阳性成纤维细胞系快速筛选新方法与普通筛选方法比较
我们利用普通筛选方法先后共转染了37种不同目的基因打靶载体至猪胎儿成纤维细胞中,经筛选后成功获得了GTKO[16]、GTKO/hCD55/hCD59三基因修饰[10]、GHRKO[15]、P53KO[17]、Leptin过表达[18]等9种不同基因编辑猪阳性成纤维细胞系,进行SCNT 后均获得了相应的基因编辑克隆猪,筛选成功率为24.3% (9/37),平均筛选周期为33.7 d。按照本研究中猪基因编辑阳性细胞快速筛选新方法先后共转染了33种不同目的基因打靶载体至猪胎儿成纤维细胞中,经筛选后成功获得IPO13KO等29种不同基因编辑猪阳性成纤维细胞系,部分经SCNT 后获得了相应基因编辑的胎儿或克隆猪,筛选成功率为87.9%(29/33) (χ2=28.38,P<0.005),平均筛选周期为18.9 d。2种筛选方法相比,筛选周期缩短约15 d(P<0.001),筛选成功率提高约4 倍(图4)。
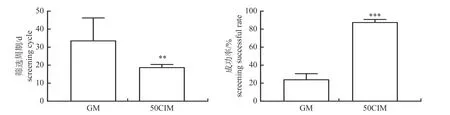
图4 新筛选方法与普通筛选方法的筛选周期与筛选成功率比较Fig.4 Comparison of screening cycle and success rate between new screening method and general screening method
3 讨论
因猪在解剖学、生理学及生化指标等方面与人极为相似,基因编辑猪在人类疾病模型、异种器官移植及比较医学等研究领域具有重要的应用价值。目前,猪尚无真正的可供基因编辑和SCNT 用的干细胞,猪胎儿成纤维细胞系仍是猪基因编辑及SCNT 供体细胞的首选。但猪成纤维细胞增殖能力有限,培养过程中易发生衰老,且在筛选过程中常需在培养基中添加G418 和Puromycin 等抗生素,导致猪成纤维细胞阳性单克隆形成周期延长,甚至可长达8~10 周。此外,通过流式细胞仪分选后,易对细胞产生物理性损伤,不利于单细胞克隆的形成和传代培养,造成猪基因编辑阳性细胞筛选的成功率及SCNT效率低下。如图5 所示:在基因编辑阳性猪胎儿成纤维细胞筛选的过程中,普通的筛选方法需将单细胞克隆增殖至24 孔板后再收集1/2的细胞量进行细胞基因型鉴定,然而猪成纤维细胞增殖能力有限,在此过程中,需对单细胞克隆进行多次的胰蛋白酶消化传代,对细胞的损伤加大,进一步导致细胞发生衰老,细胞活力变差甚至死亡。其次,因成纤维细胞的增殖具有密度依赖性,大量的单细胞克隆被用于鉴定会致使剩余细胞密度较低,不利于长期传代和扩大培养,甚至在传代的过程中造成细胞死亡,导致基因编辑阳性细胞丢失,严重制约了基因编辑阳性细胞系的筛选效率和基因编辑克隆猪的产生。
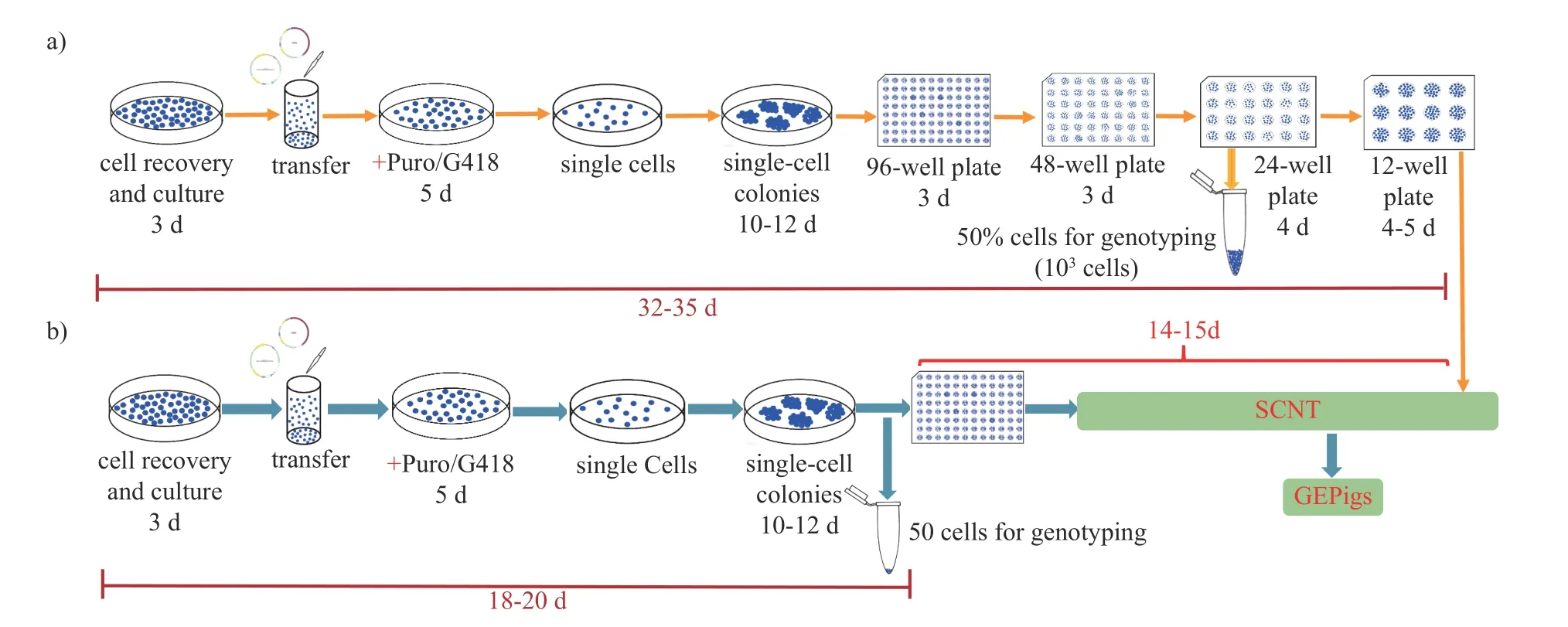
图5 基因编辑猪阳性成纤维细胞筛选示意图Fig.5 Diagram of selection of positive porcine gene-editing fibroblasts
供体细胞的质量也是影响SCNT 效率及克隆胚胎发育的重要因素。在SCNT 研究过程中,供体细胞的代次[19]、细胞分化状态[9,20]、培养方式[21-23]、同期化水平[24]及表观遗传状态[19,25]均可影响SCNT的效率。在体外短期培养的供体细胞比长期培养的供体细胞更容易进行核移植重编程,以早期代次的供体细胞进行核移植后的胚胎发育率优于晚期代次的供体细胞[26]。在山羊体细胞克隆中,随着山羊成纤维细胞传代次数的增加,细胞形态学发生变化,高代次的细胞衰老显著高于低代次细胞,且高代次供体细胞构建的重构胚的发育能力低于低代次[27]。本研究在前期大量猪基因编辑阳性细胞筛选的基础上,优化了细胞筛选的各个环节,建立了猪基因编辑阳性成纤维细胞系快速筛选方法。该方法在单细胞克隆形成的早期阶段(96 孔板中)即可对单细胞克隆进行准确计数,取50 个细胞进行基因型鉴定,鉴定为基因编辑阳性的细胞系可立即用于SCNT,从而使细胞筛选周期缩短约15 d,同时也减少了供体细胞体外培养时间和传代次数,最大限度地保证了细胞的活力,提高了SCNT 重编程的效率。但是,该方法在96 孔板中(约2 000 个细胞)取出1 000 个阳性细胞作为供体细胞进行SCNT,剩余的细胞最多还可用于3 次SCNT。若细胞用完,则需重头进行阳性供体细胞筛选。若通过外科手术从妊娠母猪中取胎儿并进行基因型鉴定后,建立相应的猪胎儿成纤维细胞系,可获得大量具备高活力的基因编辑猪阳性细胞系,从而保证充足的SCNT 供体细胞来源。总之,本研究建立的方法在缩短筛选周期的同时,还大大提高了细胞筛选的成功率,可最大限度地维持猪成纤维细胞的增殖能力,获得大量高活力的基因编辑阳性猪胎儿成纤维细胞系,为后续批量的SCNT 及相关基因功能验证提供了充足的细胞资源。
4 结论
本研究建立了一种猪基因编辑阳性成纤维细胞系快速筛选的方法,并应用该方法快速筛选获得IPO13KO等29种不同基因编辑的猪阳性细胞系;进一步以此为供体细胞进行SCNT,成功获得9 头IPO13KO克隆猪。与普通筛选方法相比,该方法促使筛选周期缩短约15 d,筛选成功率提高约4 倍,为快速产生猪基因编辑克隆猪提供了重要的理论依据和借鉴方法。
猜你喜欢
今日农业(2020年24期)2020-12-15
科学(2020年4期)2020-11-26
科学(2020年4期)2020-11-26
中西医结合心脑血管病杂志(2016年20期)2016-03-01
上海农业学报(2016年5期)2016-02-10
山东医药(2015年14期)2016-01-12
江苏大学学报(医学版)(2015年2期)2015-04-17
中国医药导报(2015年26期)2015-02-28
生殖医学杂志(2015年11期)2015-02-28
化学工业与工程(2015年1期)2015-02-10
