
LPIN1基因突变所致先天性肌病1例报道并文献复习
2021-11-03 06:35高华杰季苏琼马雪毕抓劲张清杨梦歌卜碧涛
神经损伤与功能重建 2021年10期
高华杰,季苏琼,马雪,毕抓劲,张清,杨梦歌,卜碧涛
先天性肌病是一组临床和遗传异质性肌肉疾病[1,2],其临床表现为出生时或出生后肌张力低下、肌无力、先天性骨骼畸形,伴或不伴运动发育迟滞,可有心肌、呼吸肌及眼肌异常但较少见,病情相对稳定或呈缓慢进展[3-5]。现报道1例先天性肌病并对相关进展进行文献回顾。
1 资料与方法
1.1 病例资料
患者,男,9岁7个月,主因“四肢无力9年余,加重伴右侧肢体阵发性疼痛不适3天”于2017年3月28日入院。患者自幼肢体肌力差,2岁时才能自行行走,伴明显的行走不稳,反复就医。3岁时肌活检结果提示先天性肌病可能,电镜示脂质沉积性肌病。给予营养神经、改善肌肉代谢等对症支持治疗,患者病情反复波动-好转。3天前无明显诱因逐渐出现右侧肢体阵发性疼痛,以右侧肘关节及小腿为甚,伴右肘关节曲伸受限,左侧手指间断不自主呈爪形捏合,动作笨拙,行走不利,易摔跤。既往史:患儿顺产,自幼体弱,体型偏瘦,体力较同龄人差,智力发育尚可。否认肝炎、结核等传染病接触式史,否认药物及食物过敏史。家族史:父母近亲婚配,弟弟无类似病史。
入院查体:神志清楚,吐词清晰,双瞳孔等大等圆,直径3 mm,对光反射灵敏,眼球活动自如;行走不稳,四肢肌力4级,肌张力减低,腱反射可,深浅感觉未见明显异常,病理征(-);颈软、无抵抗,克氏征(-),Gower(+)。胸部、腹部查体未见明显异常。入院前辅助检查:2011年我院右侧腓肠肌肌肉活检:HE示肌纤维大小形态基本正常,未见坏死及再生,未见核内移,未见异常空泡或包涵体,未见炎症细胞浸润,未见小血管炎症,结缔组织增生不明显;ATP未见群组,I型纤维占明显优势,约70%~80%,II型纤维较少,大小形态基本正常,未见肯定萎缩纤维,见图1。电镜有串珠样脂质沉积。2011年外院查DMD基因未见异常。

图1 患者右侧腓肠肌肌肉活检ATP酶染色(×100)
入院后辅助检查:血常规、甲状腺功能全套、甲状腺免疫全套、葡萄糖、糖化血红蛋白、凝血四项未见明显异常。心梗三项:肌红蛋白>1200 ng/mL、肌酸激酶MB型同工酶>300 ng/mL。生化全套(含血糖):谷丙转氨酶684 U/L、谷草转氨酶1479 U/L、肌酸激酶>20000 U/L、乳酸脱氢酶>1867 U/L。尿常规:茶色,红细胞29.6/μL。心电图:窦性心律,心电图正常范围。彩超心脏:心脏形态结构及瓣膜活动未见明显异常,EF67%。磁共振-大、小腿平扫:双侧股二头肌下段、双侧股直肌、双小腿腓肠肌、比目鱼肌、胫骨前肌、胫骨后肌、腓骨长短肌走行区异常信号,见图2。考虑诊断为先天性肌病。

图2 患者大、小腿肌肉核磁影像(2017年4月)
为缓解患者疼痛症状,经患者家属同意后给予甲基强的松龙120 mg静滴,1次/d,同时给予护胃、补钾、补钙等对症治疗;患者及其父母、弟弟抽血查遗传性肌病基因。基因检测结果显示:患者LPIN1基因c.357_358insCT纯合突变,见图3。第1次复查血心梗三项:肌红蛋白57.3 ng/mL、肌酸激酶MB型同工酶7.9 ng/mL;生化全套(含血糖):谷丙转氨酶312 U/L、谷草转氨酶107 U/L、肌酸激酶1291 U/L、乳酸脱氢酶1413U/L。甲基强的松龙改60 mg静滴,1次/d,余治疗不变。第2次复查血心梗三项:肌红蛋白34.8 ng/mL、肌酸激酶MB型同工酶4.5 ng/mL;生化全套(含血糖):谷丙转氨酶169 U/L、谷草转氨酶57 U/L、肌酸激酶261 U/L、乳酸脱氢酶751 U/L。激素由静脉滴注改为强的松30 mg口服,1次/d,维持治疗。激素治疗后患者肌酸激酶水平显著降低,疼痛症状明显减轻。

图3 患者家系遗传性神经肌肉病基因检测(2017年6月)
1.2 方法
收集资料并分析。通过Pubmed数据库,输入关键词“LPIN1”和“myopathy”或“Rhabdomyolysis”,收集所有相关文献(检索时间截止2021年03月15日),得到所有关于先天性肌病和LPIN1基因的相关病例报道。采用描述性统计对符合条件的所有病例从临床表现、影像学表现、病理类型和基因结果进行分析。
2 结果
共检索文献31篇(不包含综述)。共搜索到LPIN1基因相关肌病患者66例,91%为儿童,9%为成人(≥18岁)。所有患者均表现为横纹肌溶解(肌酸激酶显著增高、肌痛、血红蛋白尿),15%的患者心肌受累(其中40%猝死),呼吸肌受累未见。14%患者做过病理检查(其中57%为尸检)。所有患者均做了基因检测(其中纯合突变占72%,杂合突变占28%)。
3 讨论
先天性肌病是由遗传决定的肌肉结构蛋白缺陷引起的异质性疾病[2]。患者发病年龄早,主要表现为肌张力水平低下、肢体无力,病情相对稳定或呈缓慢进展。发病率估计约为1∶25000[6]。先天性肌病患者肌肉核磁提示肢体近端骨骼肌脂肪化,以臀大肌、股直肌外的股四头肌为重[2]。肌电图呈肌源性损害或者正常。肌酸激酶水平通常正常或升高。根据肌肉活检特征,先天性肌病通常分为5类:轴空性肌病、中心核肌病、杆状体肌病、肌球蛋白储积病和先天性肌纤维类型不均[1,7]。先天性肌病的诊断要点包括临床评估(症状、体征、详细的家族史),肌肉核磁检查,血清肌酸激酶测定,肌电图。但鉴于肌酸激酶水平通常正常或轻度升高,对于先天性肌病的诊断并不是特别有帮助,而肌电图也可以是正常的。因此,肌肉活检(包括组织学、免疫组织化学和电子显微镜检查)、基因检测作为一种新兴的检查手段越来越多的应用于临床诊断。该病例患者表现为横纹肌溶解(肌酸激酶显著增高、肌痛、血红蛋白尿症)。横纹肌溶解症的病因可分为创伤性和非创伤性因素[8-11]。其中,创伤性因素有运动过量、挤压;非创伤性因素有药物(他汀类、贝特类、中药甘草制剂)、内分泌疾病(低钾血症、糖尿病、原发性醛固酮增多症、甲状腺机能减退症等)、中毒(食物、百草枯等毒物),其他原因如感染、遗传性和自身免疫性疾病等[12,13]。
本次报告的患者自幼以肢体无力起病,此次发病伴有肌痛,茶色尿,肌酸激酶水平增高,神经电生理提示肌源性损害,肌肉核磁示双侧大、小腿肌肉异常信号,考虑提示肌炎或肌病可能,肌肉活检提示本患者考虑先天性肌病伴I型纤维优势,诊断为先天性肌病。患者LPIN1基因c.357_358insCT纯合突变(图2),为常染色体隐性遗传,主要临床表现为复发性横纹肌溶解、肌肉疼痛、肌无力等等[14],与本例患者症状相符。因此推测,LPIN1基因c.357_358insCT纯合变异可能为该受检者的致病突变。其父亲、母亲、弟弟均在LPIN1基因上检出c.357_358insCT杂合变异(图2);因此推测,患者在LPIN1基因c.357_358insCT纯合变异分别遗传自其父母(图4)。人类LPIN1基因定位于2号染色体2p25.1处,基因长度为2672 bp,编码890个氨基酸[15]。LPIN1基因最初是Langner在fld(fatty liver dystrophy)基因突变小鼠中发现的[16],Zeharia等[17]认为LPIN1的常染色体隐性突变是严重复发横纹肌溶解的原因。LPIN1主要在骨骼肌和脂肪组织中表达[15,18,19]。LPIN1在肌肉中显著表达,是一种依赖于Mg2+的磷脂酸磷酸水解酶,催化磷脂酸去磷酸化生成二酰甘油和无机磷酸盐[20]。LPIN1突变会造成多种疾病,迄今为止已有48例患者共计46个LPIN1突变位点被报道,见图5[21]。我们首次报告了LPIN1基因c.357_358insCT纯合变异见于先天性肌病。

图4 本例患者家系图
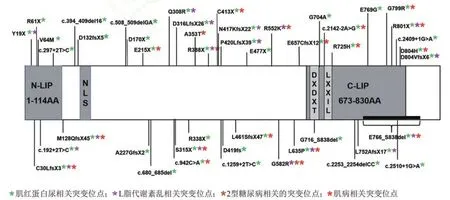
图5 LPIN1蛋白结构及已报道的人类LPIN1突变位点[21]
幼年起病、横纹肌溶解是该病例的主要特点。1例LPIN1基因纯合突变的患者,只有母亲为携带者,多次横纹肌溶解发作糖皮质激素(地塞米松)治疗有效[22]。这与糖皮质激素具有抗炎作用而且影响碳水化合物、蛋白质、脂肪代谢有关。已报道的患者均有横纹肌溶解表现,LPIN1病的预后差,有报道1/3的患者在横纹肌溶解危象中死亡[23]。
目前还没有可以治愈先天性肌病的方法,只能给予对症治疗[24,26]。如关节挛缩及脊柱侧弯者给予矫形治疗;吞咽障碍和喂养困难可予吞咽训练或鼻饲、甚至胃造瘘术以保证充足的营养支持;呼吸功能障碍者给予呼吸训练、肺部物理治疗,必要时可给予机械通气支持。因此,先天性肌病患者的治疗需要多学科共同参与,如神经科、康复医学科、儿科、整形外科、神经心理科等[25,26]。基因诊断将为患者家庭的遗传咨询提供帮助,明确的基因诊断可以通过产前诊断,降低下一胎及下一代患相同疾病的风险。目前先天性肌病尚无根治方法,但已有动物实验显示基因替代治疗将是单基因遗传先天性肌病最具希望的治疗措施[27]。
猜你喜欢
中国医院院长(2021年21期)2022-01-21
今日农业(2021年5期)2021-11-27
冰雪运动(2021年1期)2021-07-28
安徽医专学报(2020年3期)2020-12-25
医学信息(2020年12期)2020-07-27
文萃报·周二版(2019年31期)2019-09-10
科学生活(2018年5期)2018-05-31
大众健康(2016年10期)2016-12-07
医学信息(2016年29期)2016-11-28
当代体育科技(2015年8期)2015-02-27
