
1,3,5-三芳基吡唑衍生物的高效合成方法
2021-11-02 03:01解恒参丁维华赵明珠
兰州理工大学学报 2021年5期
杨 峥, 解恒参, 丁维华, 赵明珠
(1. 江苏建筑职业技术学院 江苏省生物质资源综合利用工程实验室, 江苏 徐州 221116; 2. 江苏师范大学 化学与材料科学学院, 江苏 徐州 221116)
含氮杂环化合物是一类重要的有机化合物.诸多含氮五元杂环化合物中,吡唑类化合物因其高效、低毒以及吡唑环上取代基可多方位变换的优势,表现出丰富的生物和药物活性[1-3].1-(3-三氟甲基苯基)-3-氨基吡唑啉(BW755C)[4]具有很好的抗炎疗效,是目前吡唑类化合物中药物应用的典型代表,异索威(Isolan)、Mon12800、Vaniliprod(图1)是吡唑类杀虫剂的典型代表结构单元,可用于棉花、果树和蔬菜的叶面广谱杀虫杀螨[5].
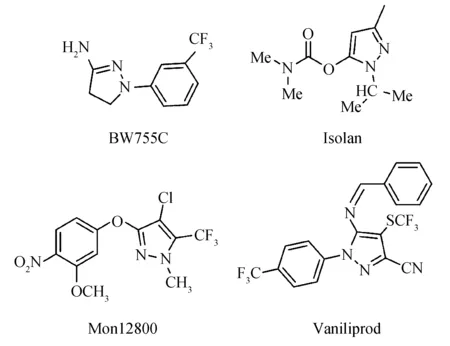
图1 含有吡唑骨架的活性分子Fig.1 Active molecules containing pyrazole skeletons
芳基吡唑结构单元具有重要的生物活性和药理特性,作为药物研发项目的一部分,一直是有机合成领域关注的焦点.Azarifar等[6]在水合硝酸铋作用下,通过吡唑啉类化合物自身氧化、芳构化合成了1,3,5-三芳基吡唑衍生物.Heller等[7]以二(三甲基硅基)氨基锂(LiHMDS)催化1,3-二酮衍生物和苯肼发生缩合反应,在甲苯溶剂中,合成了1,3,5-三芳基吡唑衍生物,收率仅为43%.该反应的缺点是使用了毒性较大的有机溶剂且收率较低.Deng等[8]在H2O-DMF体系中,使用(E)-1-(4-氯亚苄基)-2-苯基肼和(E)-5-(2-硝基乙烯基)-苯并[d][1,3]二氧杂环戊烯,回流反应4天,合成了1,3,5-三芳基吡唑衍生物,产率仅为42%.Liu等[9]报道了酰氯、端基炔烃、肼通过耦合和环缩合反应,一锅法制备了一系列1,3,5-三芳基吡唑化合物,该反应操作复杂,产物收率较低为15%,反应过程中使用了贵金属催化剂,价格昂贵.
尽管有多种合成吡唑衍生物的方法,但这些方法同时存在不少的缺点,如使用了昂贵的过渡金属催化剂,使用有毒的有机溶剂,产物收率低,反应时间较长和操作复杂等.因此,开发一个简单、高效、绿色的方法用于合成1,3,5-三芳基吡唑衍生物是当前的研究热点.
本文研究了以芳甲酰环氧乙烷和芳基肼为起始原料,在三乙胺作碱促进剂,乙醇为溶剂,微波辐射条件下(100 ℃,15 min),经原位开环/环化反应,一锅法高效合成了1,3,5-三芳基吡唑衍生物(图2),丰富了三芳基吡唑类化合物的合成方法学.
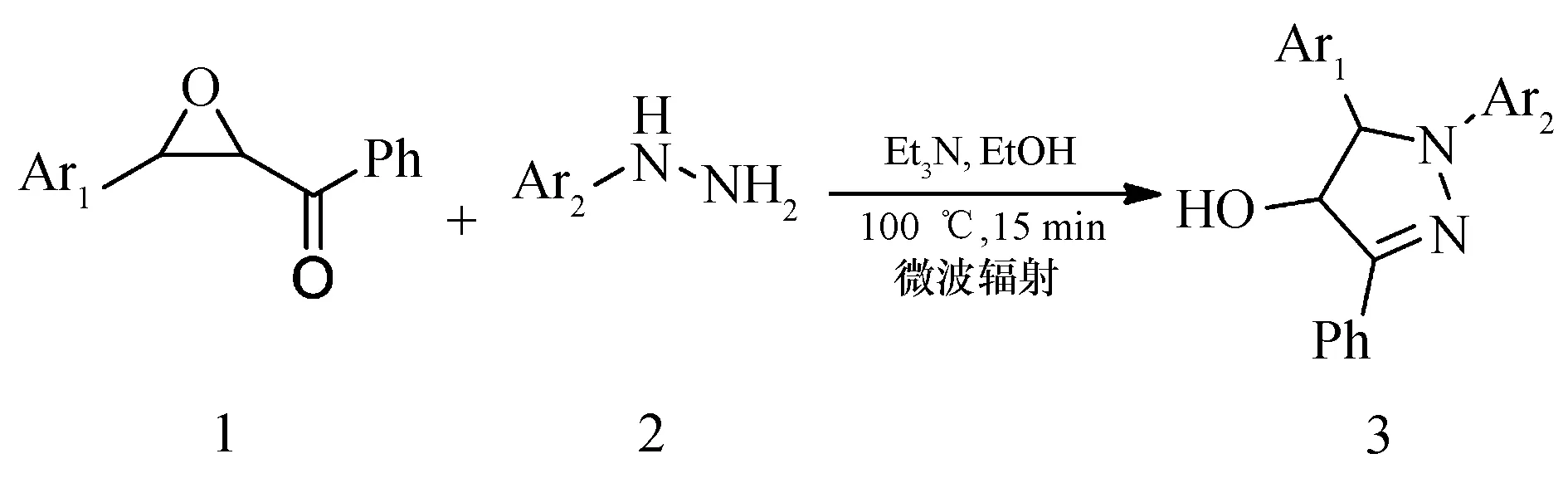
图2 1,3,5-三芳基吡唑衍生物的合成Fig.2 Synthesis of 1,3,5-triarylpyrazoles derivatives
1 实验部分
1.1 仪器与试剂
仪器:Bruker DPX 400 MHZ核磁共振波谱仪(DMSO-d6为溶剂,TMS为内标);XT-5型显微熔点测定仪(温度未经校正);Nicolet FTIR 5DX型红外光谱仪(KBr压片);micro TOF-Q Ⅱ型质谱分析仪;Siemens P4型X-射线衍射仪.
试剂:a-溴代苯乙酮、芳醛、芳基肼、三乙胺,季铵盐.药品由安耐吉化学提供,其他试剂均为市售分析纯,所有溶剂在使用前均经旋转蒸发仪精制.
原料制备:在文献[10]的基础上用改进的方法合成芳甲酰环氧乙烷,以a-溴代苯乙酮和芳醛为原料,季铵盐为相转移催化剂,乙醇为溶剂,室温搅拌(25 ℃)条件下制备原料.

1.2 合成方法
1,3,5-三芳基吡唑衍生物的合成通法:在10 mL专用的微波反应管中,顺序加入芳甲酰环氧乙烷(1.0 mmol)、芳基肼(1.0 mmol)、三乙胺(1.0 mmol)和乙醇(2.0 mL).待固液体系充分混合均匀后,设置反应温度100 ℃,反应时间15 min(最大功率为200 W).TLC跟踪反应进程,至芳甲酰环氧乙烷反应完全后,将反应器取出冷却至室温.随后加入30 mL水,乙酸乙酯萃取(3次×4 mL),经粗硅胶柱层析(200~300目)(V(乙酸乙酯)∶V(石油醚混合物)=1∶5为洗脱剂),得到一系列1,3,5-三芳基吡唑衍生物.
1.3 表征数据
3a 浅黄色固体;收率:73%;熔点:124~125 ℃;红外:3 312、1 706、1 532、1 424、1 312、1 150、1 020 cm-1;氢谱(400 MHz,DMSO-d6)、7.85(d,J=7.2 Hz,2H),7.52~7.26(m,8H),7.19(d,J=7.2 Hz,2H),7.01(d,J=8.4 Hz,2H),6.53(s,1H),5.21(s,1H),5.04(s,1H);质谱(ESI):m/z理论值C21H16BrN2O:391.044 6 [M-H]-;实际值:391.043 0.
3b 黄色固体;收率:59%; 熔点:124~125 ℃;红外:3 352、1 656、1 596、1 495、1 323、1 136、1 033 cm-1;氢谱(400 MHz,DMSO-d6):8.32(s,1H),8.05(d,J=7.6 Hz,2H),7.82(d,J=8.0 Hz,2H),7.58~7.42(m,2H),7.32(d,J=2.0 Hz,3H),7.18(d,J=7.6 Hz,1H),7.03(d,J=7.6 Hz,2H),6.85(d,J=7.2 Hz,1H),5.13(s,1H),4.84(s,1H);质谱(ESI):m/z理论值C21H16FN2O:331.124 7 [M-H]-;实际值:331.124 2.
3c 黄色固体;收率:78%;熔点:116~117 ℃;红外:3 356、1 734、1 612、1 408、1 395、1 122、1 076 cm-1;氢谱(400 MHz,DMSO-d6):7.84(d,J=7.2 Hz,2H),7.41(t,J=7.6 Hz,2H),7.36~7.32(m,3H),7.28~7.16(m,5H),7.06(d,J=7.6 Hz,2H),6.75(s,1H),6.44(d,J=7.6 Hz,1H),5.16(d,J=2.8 Hz,1H),5.03(d,J=4.8 Hz,1H);质谱(ESI):m/z理论值C21H17N2O:313.134 1 [M-H]-;实际值:313.134 0.
3d 黄色固体;收率:61%;熔点:106~107 ℃;红外:3 268、1 664、1 526、1 477、1 335、1 251、1 098 cm-1;氢谱(400 MHz,DMSO-d6):7.84(d,J=7.2 Hz,2H),7.41(t,J=7.6 Hz,2H),7.34(t,J=6.4 Hz,3H),7.28~7.20(m,3H),7.07~7.01(m,2H),6.76(d,J=8.0 Hz,1H),6.58(d,J=7.2 Hz,1H),6.46(d,J=7.6 Hz,1H),5.16(s,1H),5.01(d,J=7.6 Hz,1H),2.23(s,3H);质谱(ESI):m/z理论值C22H19N2O;327.149 7 [M-H]-;实际值:327.152 1.
3e 浅黄色固体;收率:61%;熔点:139~140 ℃;红外:3 316、1 654、1 524、1 388、1 264、1 108、1 033 cm-1;氢谱(400 MHz,DMSO-d6):7.75(d,J=7.2 Hz,2H),7.52~7.36(m,7H),7.30(s,1H),7.05(s,1H),6.94(t,J=7.6 Hz,1H),6.79(d,J=8.0 Hz,1H),6.54(d,J=7.2 Hz,1H),3.63(s,1H),3.52(s,1H),2.17(s,3H);质谱(ESI):m/z理论值C22H19N2O:327.149 7[M-H]-;实际值:327.152 1.
3f 黄色固体;收率:69%;熔点:135~136 ℃;红外:3 352、1 688、1 542、1 402、1 298、1 113、1 048 cm-1;氢谱(400 MHz,DMSO-d6):7.20(s,1H,ArH),8.03(d,J=7.2 Hz,2H),7.73(d,J=8.0 Hz,2H),7.58(t,J=7.6 Hz,2H),7.50(d,J=2.0 Hz,3H),7.38~7.34(m,3H),7.28(d,J=7.6 Hz,1H),7.08(d,J=7.6 Hz,2H),6.92(d,J=7.2 Hz,1H),4.86(s,1H),4.19(s,1H);质谱(ESI):m/z;理论值C21H16ClN2O:347.095 1 [M-H]-;实际值:347.092 3.
3g 浅黄色固体;收率:72%;熔点:132~133 ℃;红外:3 304、1 716、1 522、1 308、1 256、1 178、1 044 cm-1;氢谱(400 MHz,DMSO-d6):7.85(d,J=7.2 Hz,2H),7.64(t,J=7.2 Hz,2H),7.55~7.42(m,3H),7.28~7.20(m,3H),7.07~7.01(m,2H),6.98(d,J=8.0 Hz,2H),6.53(d,J=7.6 Hz,1H),5.24(s,1H),5.03(d,J=7.2 Hz,1H);质谱(ESI):m/z理论值C21H16BrN2O:391.044 6[M-H]-;实际值:391.045 5.
3h 浅黄色固体;收率:62%;熔点:118~119 ℃;红外:3 298、1 674、1 556、1 324、1 202、1 194、1 088 cm-1;氢谱(400 MHz,DMSO-d6):7.85(d,J=7.2 Hz,2H),7.51(d,J=2.0 Hz,1H),7.39(t,J=7.2 Hz,2H),7.33(d,J=7.2 Hz,1H),7.25~7.21(m,2H),7.08(d,J=6.4 Hz,1H),7.03(d,J=8.0 Hz,2H),6.85(d,J=8.0 Hz,2H),5.56(s,1H),5.13(d,J=6.0 Hz,1H);质谱(ESI):m/z理论值C21H15Cl2N2O:381.056 1 [M-H]-;实际值:381.056 6.
2 结果与分析
2.1 条件优化
如表1所示,选用3-苯基-2,3-环氧-1-苯基丙酮1a(1.0 mmol)和对溴苯肼2a(2.0 mmol)为模板反应.在乙醇(EtOH)作溶剂,80 ℃微波条件下,优化筛选反应的最佳条件.

表1 化合物3a的条件优化Tab.1 Optimization for the synthesis of compound 3a
依据碱性强弱,顺次选用乙醇钠(EtONa)、氢氧化钠(NaOH)、碳酸铯(Cs2CO3)、碳酸钾(K2CO3)和三乙胺(Et3N)作为碱促进剂.结果表明:当用碱性较强的EtONa和NaOH为碱促进剂时,所得目标产物3a的收率相对较低.调整碱性强弱使用Cs2CO3和K2CO3进行反应时,分别以32%和48%的分离收率得到相应的目标产物3a.使用碱性较弱的Et3N作为碱促进剂时,产物3a的收率可达到60%.
选用Et3N为碱促进剂,分别以乙二醇(Ethylene Glycol)、N,N-二甲基甲酰胺(DMF)、乙腈(MeCN)做溶剂,80 ℃ 微波辐射条件下,产物3a的收率均不甚理想(痕量收率≤5%).
选用Et3N为碱促进剂,EtOH做溶剂,温度由80 ℃升至100 ℃时,所得目标产物3a的收率提高至72%.继续升高反应温度至120 ℃,产物收率稍有下降.
综上可知:该反应的最优反应条件为Et3N作碱性促进剂,EtOH做溶剂,100 ℃微波辐射条件下反应15 min.

2.2 底物拓展
芳基肼2的底物拓展.当芳甲酰环氧乙烷1芳环上无取代基(Ph)时,在最优条件下考察芳基肼2的底物普适性.如图3所示,芳基肼2中Ar2的取代位无论是电中性基团(Ph),供电子基团(3—CH3),还是(强)吸电子基团(4—F、4—Br),均不影响反应的进行.反应不受取代基位置的影响,间位取代的芳基肼同样给出很好的目标产物收率(59%~78%).
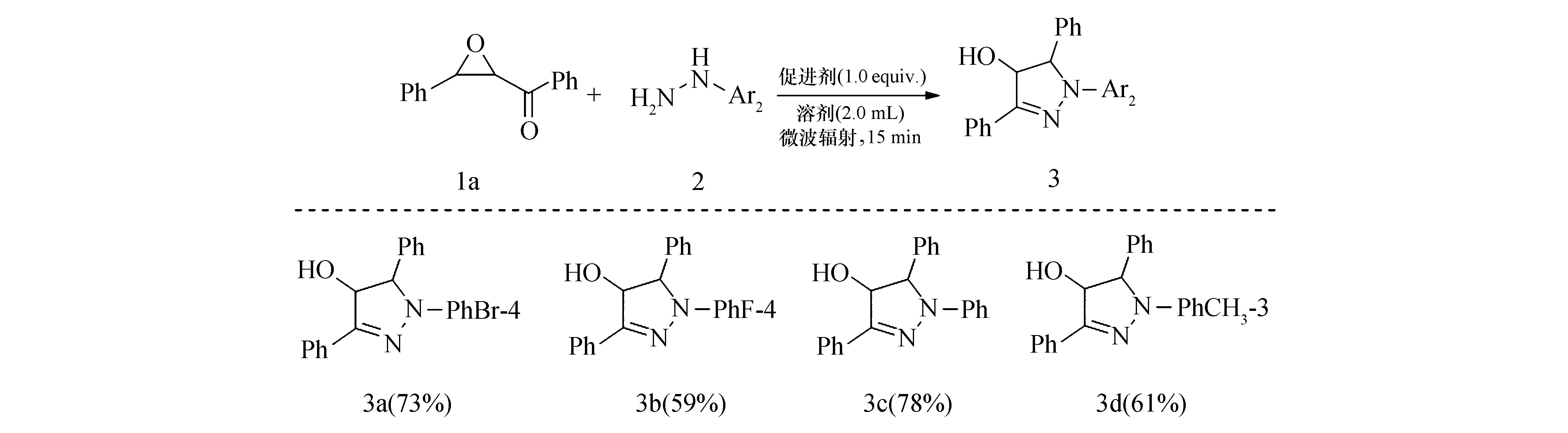
图3 芳基肼2的底物拓展
芳甲酰环氧乙烷1的底物拓展.当芳基肼2芳环上无取代基(Ph)时,在最优条件下考察芳甲酰环氧乙烷1的底物普适性.如图4所示,芳甲酰环氧乙烷1中Ar1的取代位无论是供电子基团(4—CH3),还是吸电子基团(4—Cl、4—Br、2,4—Cl2),均不影响反应的进行.以较高收率得到目标产物,且取代基位置变化不会影响该反应的进行(65%~72%).
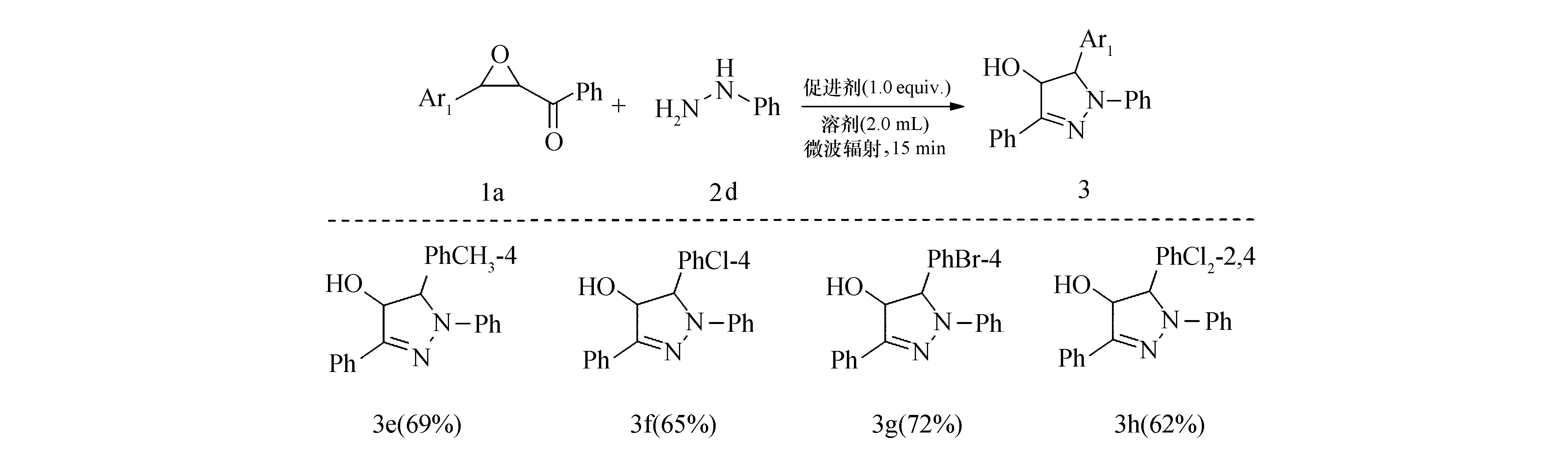
图4 芳甲酰环氧乙烷1的底物拓展Fig.4 Scope of 2,3-epoxypropan-1-ones 1
由此可见,该串联环化反应不受空间位阻及电子效应的影响.
2.3 单晶测试
对产物3a进行单晶X-射线衍射分析,图5为分子结构透视图,表2为晶体学参数.
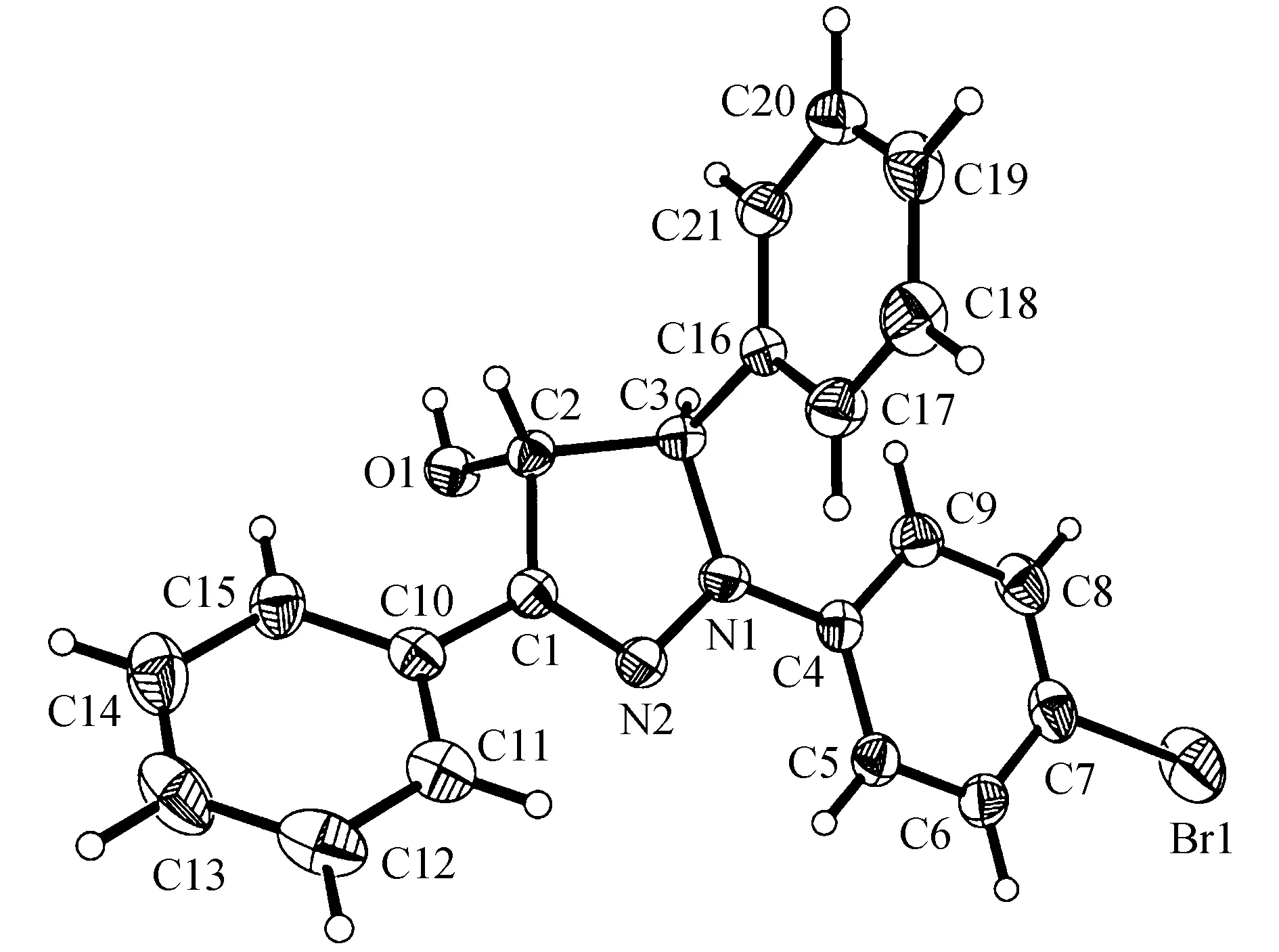
图5 化合物3a的分子结构透视图

表2 化合物3a的晶体学参数Tab.2 Crystallographic data of compound 3a
2.4 反应路径
芳甲酰环氧乙烷1与芳基肼2进行亲核加成,形成中间体A,在碱性条件下,芳基肼上的仲氨进攻环氧乙烷并开环,形成氧负离子,经分子间成环反应,形成中间体C,去质子化,得到目标产物3,如图6所示.

图6 可能的反应路径Fig.6 Plausible mechanism
3 结论
芳甲酰环氧乙烷和芳基肼在碱(Et3N)作用下,通过原位开环/环化反应,构建了吡唑杂环骨架.该反应具有条件温和、反应时间短、收率优良、操作简单、溶剂绿色友好、后处理方便等优点.表征数据翔实、产物结构新颖、官能团兼容性较好.
致谢:本文得到江苏建筑职业技术学院博士专项科研启动基金(JYJBZX20-09)和江苏建筑职业技术学院实践创新专项项目(JYSCZ20-14)的资助,在此表示感谢.
猜你喜欢
家庭医药(2022年1期)2022-01-18
今日农业(2021年2期)2021-11-27
今日农业(2020年23期)2020-12-31
初中生学习指导·中考版(2020年7期)2020-09-10
润滑油(2019年6期)2019-11-29
教育与职业(下)(2019年7期)2019-08-15
中国管理信息化(2018年7期)2018-05-27
今日农药(2017年2期)2017-03-24
农村农业农民·B版(2016年5期)2016-05-14
山东工业技术(2016年7期)2016-04-08
