
甘草饮片指纹图谱的建立及主要成分的含量测定
2021-11-01 05:33汪洋黄宇飞金文芳王国凯朱月健葛德助刘劲松安徽中医药大学药学院中药研究与开发安徽省重点实验室合肥00安徽普仁中药饮片有限公司安徽亳州6800安徽济人药业有限公司安徽亳州6800
中南药学 2021年10期
汪洋,黄宇飞,金文芳,王国凯,朱月健,葛德助,刘劲松*(. 安徽中医药大学药学院 中药研究与开发安徽省重点实验室,合肥 00;. 安徽普仁中药饮片有限公司,安徽 亳州 6800;. 安徽济人药业有限公司,安徽亳州 6800)
甘草为豆科植物甘草Glycyrrhiza uralensisFisch.、胀果甘草Glycyrrhiza inflataBat. 或光果甘草Glycyrrhiza glabraL.的干燥根和根茎,味甘性平,作用温和[1-2],具有抗炎、抗肿瘤、治疗心脑血管疾病和抗病毒等作用[3-6]。
随着中药指纹图谱的不断发展和应用,指纹图谱和多成分含量测定方法已经能提供丰富的指标成分和药效成分信息,可以较为全面地反映中药材饮片的质量,近年来已被广泛用作中药材的质量控制方法[7-8]。甘草作为一种大宗药材,已有多位学者对其指纹图谱或含量测定进行过研究[9-10],这些研究也为甘草质量标准的控制奠定了基础。但是通过对文献的研究发现,多数学者都是研究同一产地或者同一品种甘草药材的指纹图谱[11-13],对甘草饮片质量的研究较少。同时,由于市场中药材来源的不确定性,会导致甘草饮片质量参差不齐。为了能更有效地控制甘草药材饮片的质量,区分甘草饮片的品质,本研究采用高效液相色谱法建立了不同地区、不同市场和不同产地的多批次甘草饮片的指纹图谱,为质量标准的建立提供了依据。
1 仪器与试药
Waters 1525高效液相色谱仪(美国Waters 1525);超纯水处理系统(美国PALL CascadaTMⅡⅠ5);超声波清洗机(昆山市超声仪器有限公司 KQ2200);分析天平(瑞士 METTLER TOLEDO AB135-S);乙腈(色谱纯,瑞典Oceanpak公司);磷酸(分析纯,天津市大茂化学试剂厂);0.22 μm微孔滤头(美国沃特世);甘草酸铵(批号:CFN99153,纯度≥98.00%)、甘草苷(批号:CFN99154,纯度≥98.00%)(武汉天植生物技术有限公司);30批甘草饮片分别采集于不同省市,由安徽中医药大学俞年军教授鉴定为豆科植物甘草(Glycyrrhiza uralensisFisch.),来源见表1。
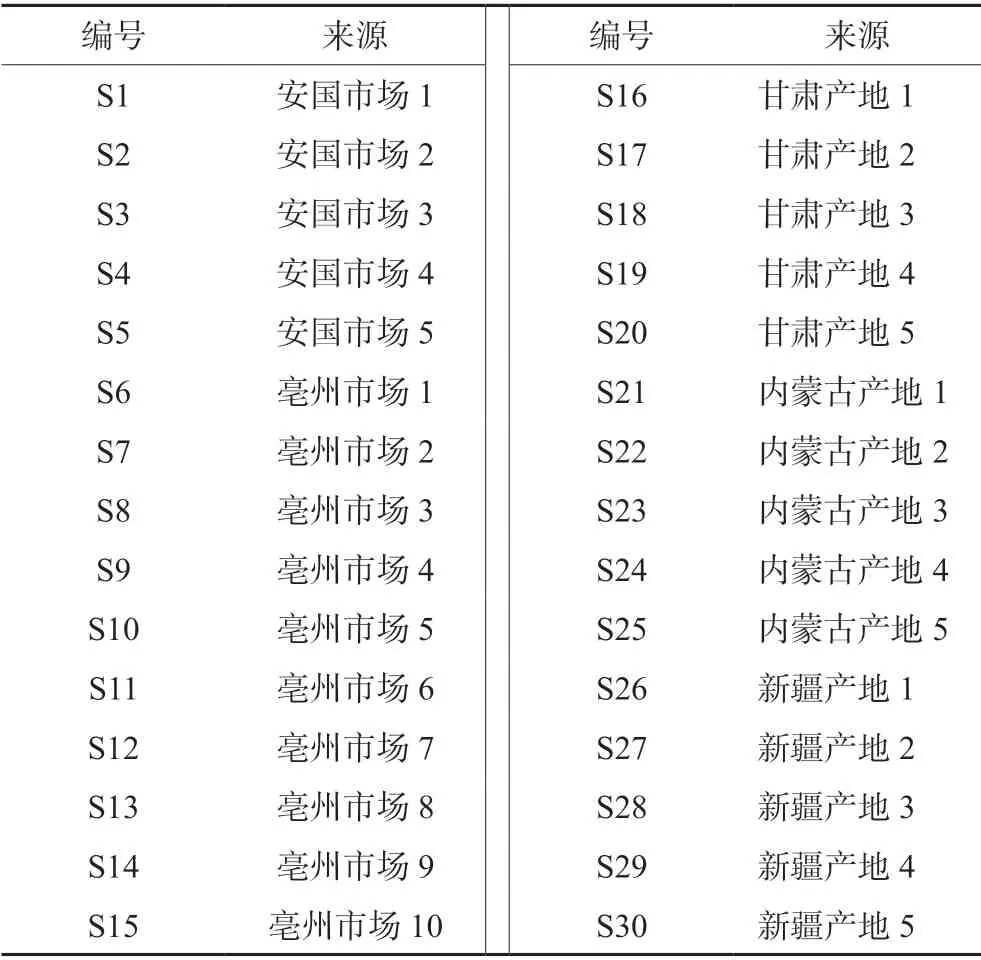
表1 样品来源Tab 1 Sample source
2 方法与结果
2.1 色谱条件
色谱柱为WondaSil C18Superb(4.6 mm×250 mm,5 μm);流动相为乙腈(B)-0.1%磷酸水(A),梯度洗脱(0~15 min,20%~30%B;15~25 min,30%~32%B;25~30 min,32%~43%B;30~43 min,43%~44%B;43~48 min,44%~90%B;48~55 min,90%~20%B);流速1 mL·min-1;柱温30℃;检测波长254 nm;进样体积20 μL。
2.2 供试品溶液的制备
取药材适量,粉碎(过3号筛),精密称取2.0 g于具塞锥形瓶中,加入70%的乙醇50 mL,称重,超声30 min,放凉后用70%的乙醇补重,摇匀后离心,取上清液过0.22 μm的微孔滤膜,即得。
2.3 对照品溶液的制备
精密称取甘草苷和甘草酸铵对照品10.00 mg,置于5 mL的量瓶中,加入70%的乙醇充分振荡溶解,定容到刻度,得到单个质量浓度为2.00 mg·mL-1的对照品溶液。精密移取适量对照品溶液置同一量瓶中混合,定容,即得混合对照品溶液。
2.4 多成分含量测定
2.4.1 线性关系考察 各取适量对照品溶液进行混合,使混合溶液中甘草苷的浓度分别为0.025、0.05、0.15、0.20、0.30、0.40 mg·mL-1;甘草酸的浓度分别为0.10、0.20、0.40、0.60、0.80、1.00 mg·mL-1。进样测定,以峰面积为纵坐标(X),质量浓度为横坐标(Y)建立线性方程,结果甘草苷标准方程为Y=1.31×107X-2.61×105(r=0.9996);甘草酸为Y=6.32×106X-1.75×105(r=0.9997)。
2.4.2 精密度试验 连续进样6次同一供试品溶液(S7),计算甘草苷和甘草酸的峰面积的RSD分别为1.7%和0.86%,证明仪器精密度良好。
2.4.3 稳定性试验 分别在0、2、4、8、12、24 h后进样同一批供试品溶液(编号:S7),得甘草苷和甘草酸峰面积的RSD分别为0.84%和1.7%,表明溶液在24 h内稳定。
2.4.4 重复性试验 进样检测同一批供试品溶液6次(S7),计算甘草苷和甘草酸含量的RSD分别为1.6%和1.5%,说明方法重复性良好。
2.4.5 加样回收试验 精密称取0.50 g同一批药材粉末6份(S7),分别加入0.12 mg甘草苷和0.55 mg甘草酸铵的对照品,制备供试品溶液,进样测定,甘草苷和甘草酸加样回收率的平均值分别为100.34%和100.52%,其RSD值分别为0.98%和0.88%,表明此方法准确可行,回收率良好。
2.4.6 含量测定结果 将甘草饮片粉末制备成供试品溶液,进样测定,外标法计算样品中甘草酸和甘草苷成分的百分含量,结果见表2。S6、S8、S10、S22的甘草酸的含量高于其他批次,其甘草苷的含量也高于大部分批次。
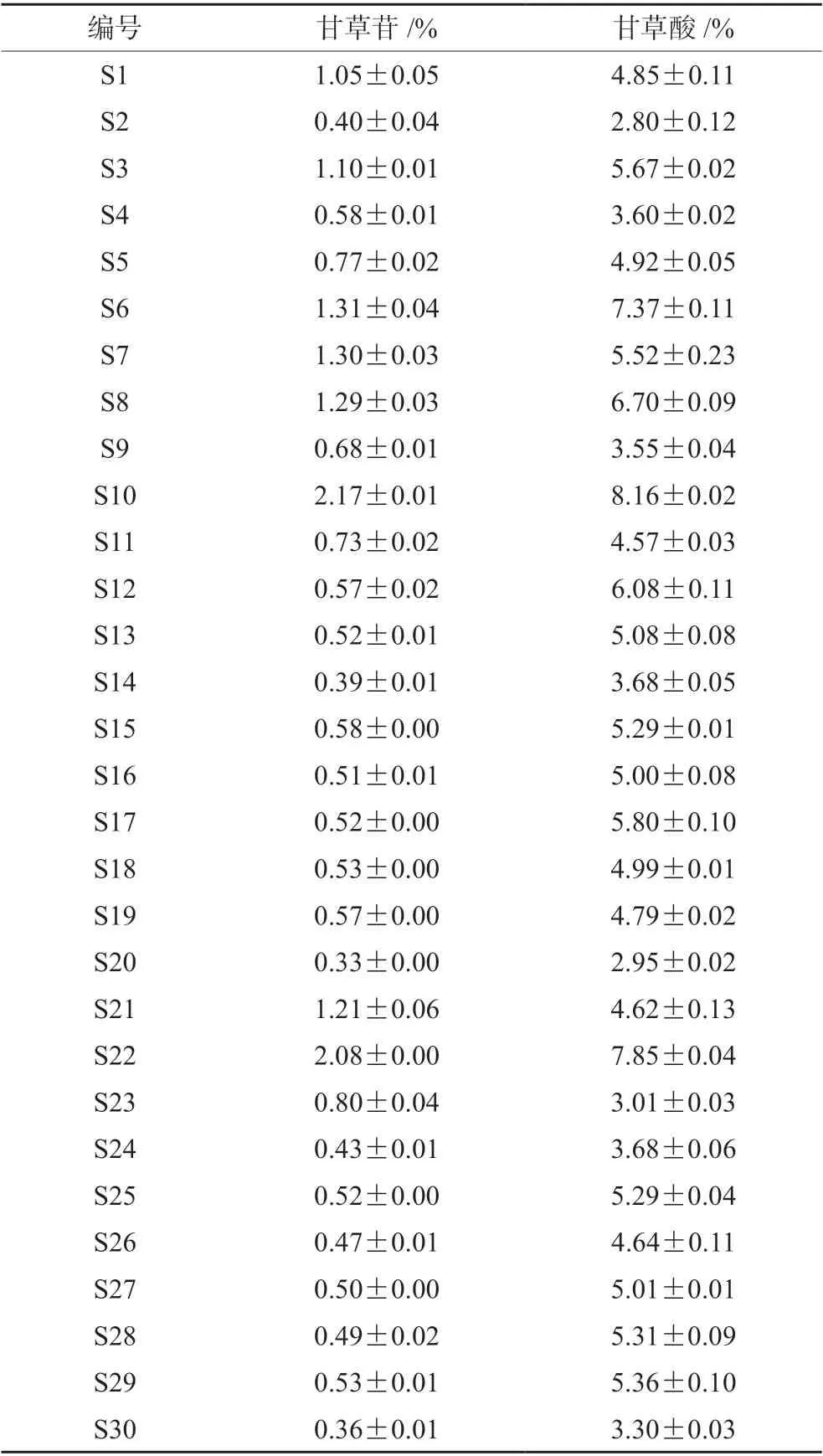
表2 30批甘草药材含量测定结果(n=2)Tab 2 Determination of 30 batches of licorice materials (n=2)
2.5 指纹图谱
2.5.1 指纹图谱的建立及特征峰的指认 按“2.2”项下方法制备(S1~S30)供试品溶液,进样检测。将混合对照品溶液色谱图与S1号色谱图进行对比(见图1),发现2号峰与13号峰的保留时间与混合对照品一致,并通过光谱对比,确定2号峰为甘草苷,13号峰为甘草酸。
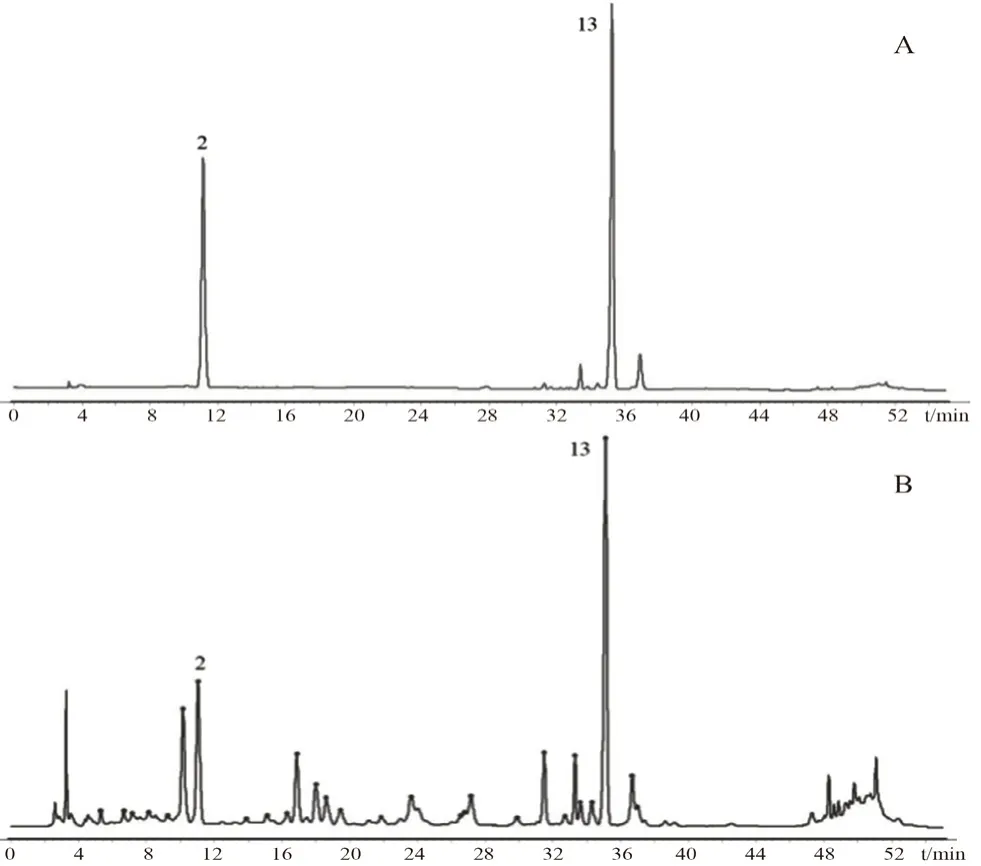
图1 混合对照品溶液(A)和样品S1共有模式(B)的色谱图Fig 1 Chromatogram of the mixed standard solution(A)and S1 common mode(B)
2.5.2 相似度评价 采用2012版的中药指纹图谱软件对30批药材图谱进行多点校正,校正完成后选择Mark峰匹配,匹配后30批药材的叠加色谱图见图2,选择S1号色谱图作为参照图谱,进行相似度计算,结果相似度均在0.941~0.999。
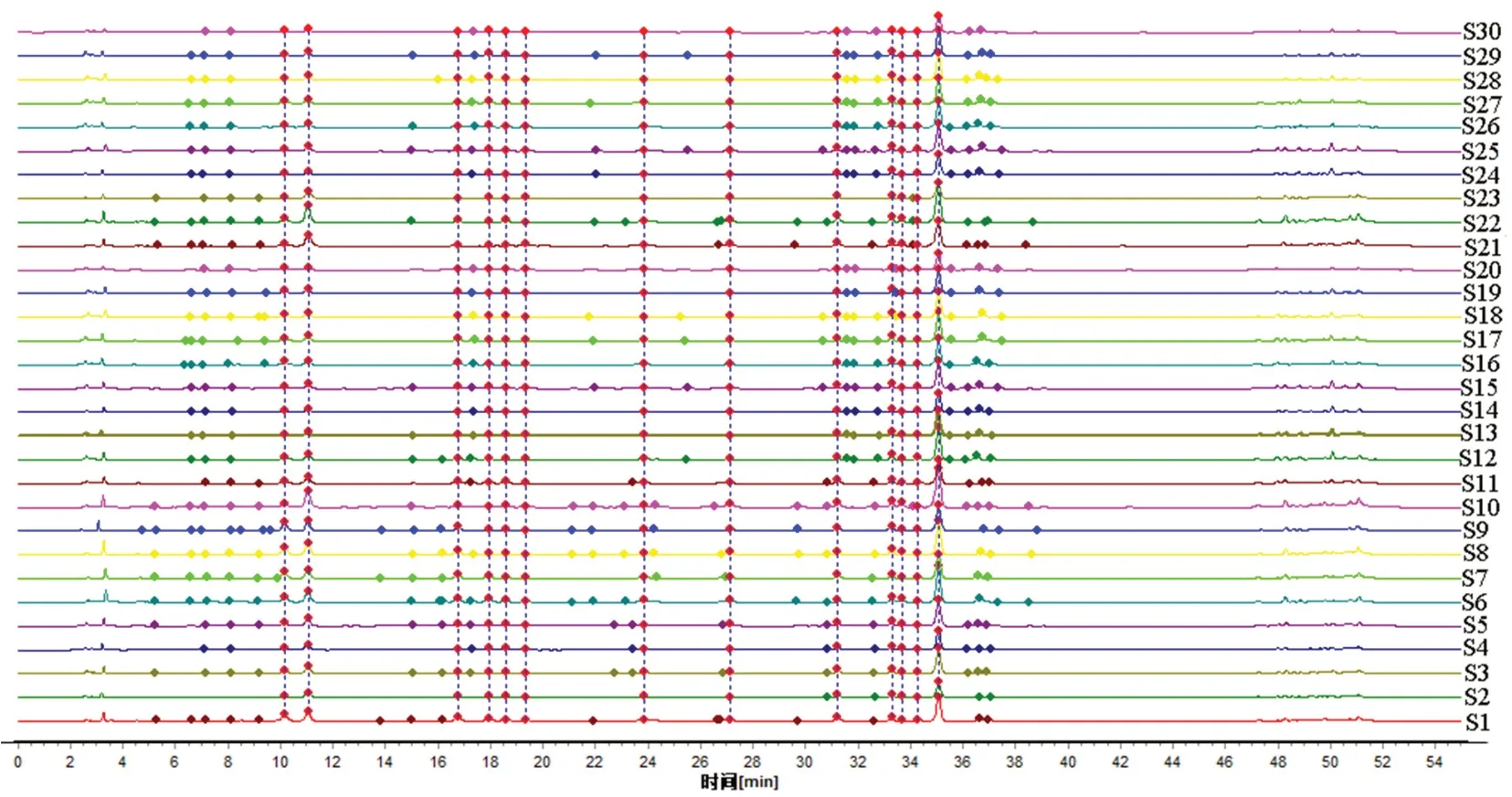
图2 30批甘草药材叠加色谱图Fig 2 Chromatograms of 30 batches of licorice materials
2.6 多元统计学分析
2.6.1 聚类分析 使用SPSS 23.0软件,采用系统聚类法,以各共有峰峰面积为变量对药材样品进行聚类分析[14-16],结果见图3。当把范围设置为10时,采用聚类分析法可将30批甘草药材聚为2类,S6、S8、S10、S22为Ⅰ类,S1~S5、S7、S9、S11~S21、S23~S30为Ⅱ类。结果内蒙古产地与亳州市场的药材聚为了一类,我国市面上的大部分甘草品种为乌拉尔甘草[17],可能是因产地内蒙古和亳州市场的药材为同一品种。
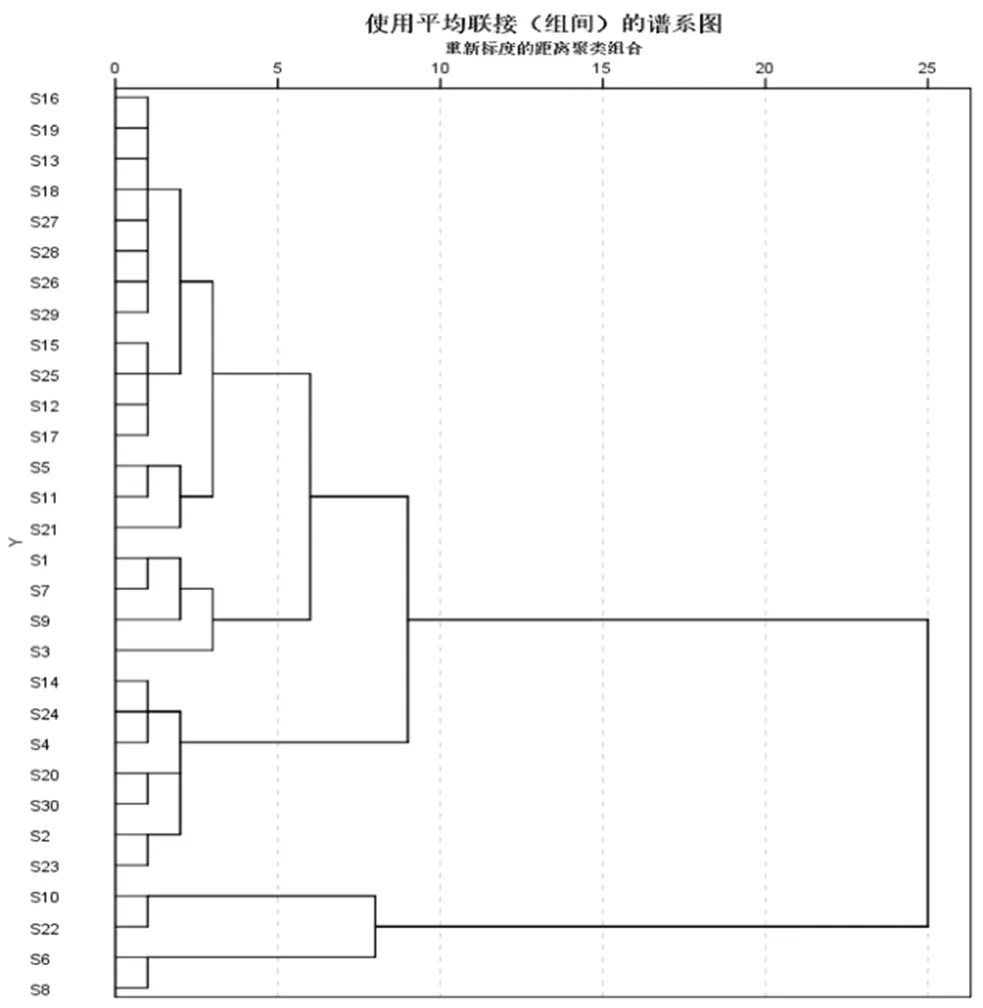
图3 30批甘草药材聚类分析树状图Fig 3 Clustering analysis dendrogram of 30 batches of licorice materials
2.6.2 主成分分析 采用SPSS 23.0软件对30批不同批次甘草药材的13个共有峰的数据进行主成分分析,计算得KMO值大于0.5,存在显著差异(P<0.05)。计算主成分特征值和累计贡献率,以特征值大于1为标准,得到前3个主成分因子的特征值分别为7.010、3.049、1.048,特征值的大小代表了矩阵正交化之后所对应特征向量对于整个矩阵的贡献程度,前3个主成分因子的累计方差贡献率为85.43%(>85.00%),说明这3个主成分代表了甘草药材13个共有峰85.43%的信息量,具有很好的代表性。主成分分析碎石图在<3时陡峭,>3时平缓(见图4),说明主成分因子1、2、3可以反映甘草药材指纹图谱共有峰的大部分信息。

图4 主成分分析碎石图Fig 4 PCA gravel diagram
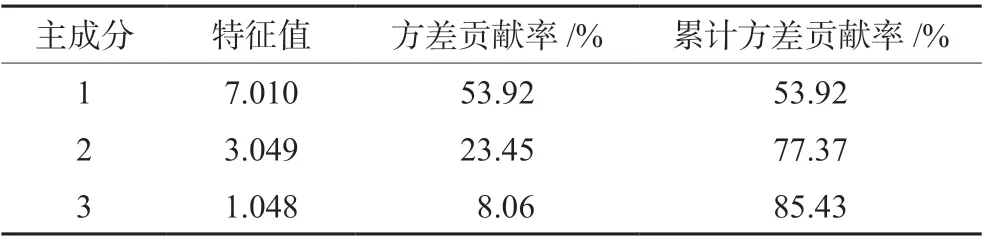
表4 主成分特征值和及方差Tab 4 Principal component eigenvalue sum and variance
2.6.3 正交偏最小二乘判别分析 分别将30批药材的共有峰面积和不同产地的共有峰面积数据导入SIMCA 14.1软件,进行有监督的OPLSDA建模分析,生成的OPLS-DA得分散点图分别为图5和图6,其预测模型的Q2分别为0.556和0.835,均大于0.5,说明建立的预测模型有效。30批药材的OPLS-DA得分散点图中发现S6、S8、S10、S22的点在二、三象限,其余各批次的点大部分都聚集在一、四象限中。不同产地的得分图中,不同产地分别聚集在不同的象限中,均有明显的区分。
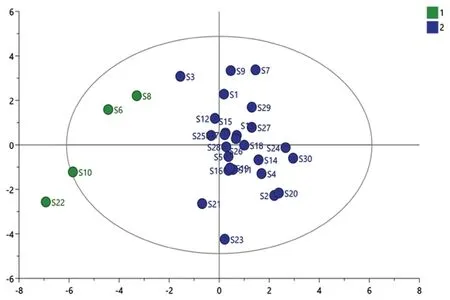
图5 OPLS-DA得分散点图Fig 5 OPLS-DA’s dispersion plots
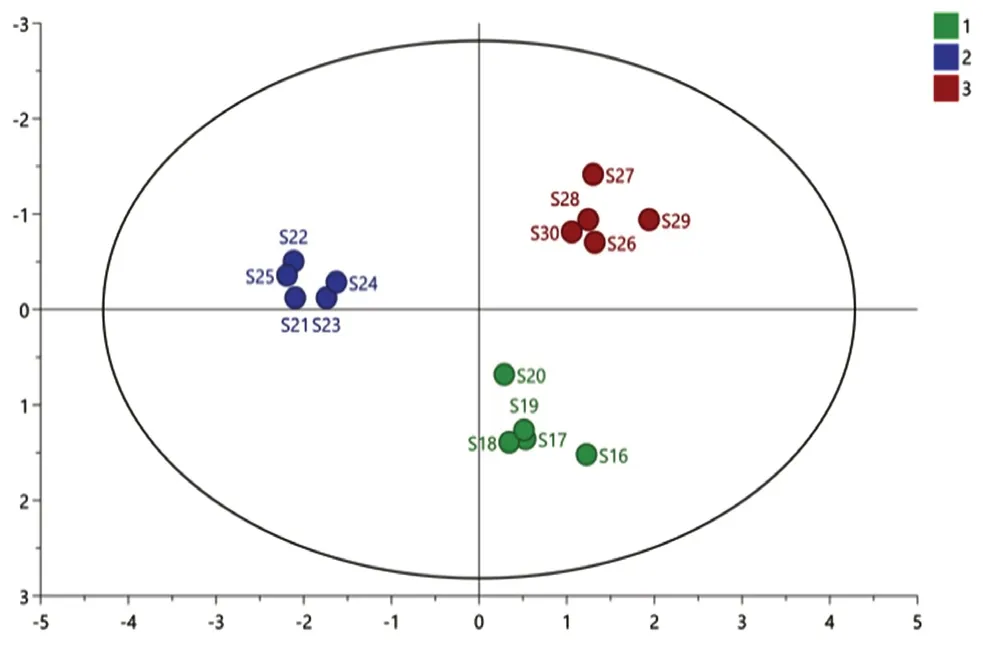
图6 不同产地OPLS-DA得分散点图Fig 6 Dispersion plots of OPLS-DA from different origins
3 讨论
本试验前期通过对甘草指纹图谱相关文献的考察,选取254 nm作为检测波长[18]。分别考察了几种不同的流动相体系(乙腈-0.05%磷酸水、乙腈-0.1%磷酸水、甲醇-0.05%磷酸水和甲醇-0.1%磷酸水),结果表明乙腈-0.1%磷酸水体系的基线更平稳,重现性最好。分别考察了不同的提取方式(超声、回流),提取溶媒(甲醇、乙醇、水),提取溶剂的浓度(30%和70%)、取样量(0.20、2.00、4.00 g)和提取时间(20、30、40 min),通过对各色谱图的对比,发现回流和超声提取的峰形差别不大,但是超声提取的方式更简单方便,故选择超声提取;不同的提取溶媒中,水提取的有效成分较少,甲醇和乙醇提取的有效成分较多,色谱图相差不大,故选择乙醇作为提取溶媒;对不同取样量进行考察,发现2.00 g和4.00 g的色谱图差别不大,但明显优于0.20 g药材的色谱图,综合考虑取样量选择2.00 g;在提取溶剂浓度的考察中,70%的浓度明显优于30%;在对提取时间的考察时,发现提取30 min后的色谱图变化不大,但有效组分的含量明显高于20 min。
本试验采用HPLC梯度洗脱建立了甘草饮片的指纹图谱,对30批次甘草饮片的HPLC指纹图谱进行分析,得到13个共有峰,软件分析其相似度均在0.9以上。通过聚类分析和主成分分析对比,发现30批次甘草饮片的HPLC指纹图谱聚类分析垂直树状图与OPLS-DA得分散点图的分类结果一致,说明了此次创建的指纹图谱方法可靠。不同产地的药材分别聚集在不同的象限内,有明显的区别。对亳州市场的10批饮片的含量进行对比,发现差别较大,这也表明在同一市场中的药材饮片的质量参差不齐,品质不一,对其进行质量控制十分必要。
猜你喜欢
基层中医药(2021年3期)2021-11-22
世界科学技术-中医药现代化(2021年5期)2021-11-05
中老年保健(2021年9期)2021-08-24
中成药(2019年12期)2020-01-04
中国卫生标准管理(2015年4期)2016-01-14
西南医科大学学报(2016年4期)2016-01-03
中国当代医药(2015年33期)2015-03-01
中国药业(2014年20期)2014-05-17
中医研究(2014年2期)2014-03-11
中国现代中药(2012年6期)2012-10-30
